References - Proceedings of the Royal Society B

Electronic Supplementary Material
Mosquito rearing and tissue generation
The experimental design, tissue and RNA collection are similar to the methods in
[1].
Six cohorts of a laboratory F
9
generation from an Ae. albopictus strain founded from at least
300 individuals collected in Manassas, VA were hatched and reared at 16h light : 8h dark, 21
C, and approximately 80% relative humidity until the pupal stage as described in
[2,3]
. Upon pupation, we established three replicate cohorts (biological replicates) of
~500 individuals each in 9.46 l adult cages under a diapause-inducing photoperiod (D; 8 h light: 16 h dark), and three cohorts under a non-diapause-inducing photoperiod (ND; 16 h light: 8 h dark). Females were allowed to blood-feed on a human host 10-13 days after eclosion. Eggs were collected over the course of a 6-hour oviposition period on three successive days for all biological replicates and treatments, which was necessary to provide sufficient material for library construction. On each day, we placed a small brown jar half-filled with ca.
50 ml of dI water and lined with unbleached seed germination paper into each cage 4 hours after lights on, and removed the jar after 6 hours. Oviposition cups were placed into cages within ~1 minute of each other, and embryo age range was counted from the time of oviposition cup placement to removal; mosquito eggs are fertilized at oviposition. The egg paper from each cage was divided equally to provide 3 day-old (“3d pov”, 72-78 hr) and 6 day-old (“6d pov”, 135-141hr) embryos for RNA extraction. All eggs were stored in the same cabinet (D photoperiod) to reduce cabinet-specific effects, and because photoperiod at the egg stage does not trigger diapause in Ae. albopictus
[4]
. In addition, embryos were reserved from each paper to assess developmental stage and diapause incidence (see below).
RNA preparation and sequencing
Individual RNA extractions were performed for each oviposition date, biological replicate, and photoperiod treatment. Embryos were ground in TRI® Reagent (Sigma
Aldrich, St. Louis, MO) at 72-73 hours (3d) or 135-136 hours (6d) after egg paper removal, resulting in 12 RNA samples (2 photoperiod treatments x 2 developmental stages x 3 biological replicates). RNA extraction and isopropanol precipitation were then performed according to manufacturer’s instructions, followed by a DNase treatment with
Turbo-DNAfree (Applied Biosystems/Ambion, Austin, TX, USA). RNA collected on successive days was pooled within each replicate and treatment after quality assessment on an RNA chip (Bioanalyzer 2100, Agilent Technologies, Santa Clara, CA).
Data preparation and read cleaning
We performed several pre-assembly steps to remove contaminated or low-quality reads from the Illumina libraries. First, we used sshaha2
[5]
to identify and remove reads with matches to the NCBI UniVec Core database ( ftp://ftp.ncbi.nih.gov/pub/UniVec/ ; accessed August 10 th
2010) and Ae. albopictus rRNA sequence (GenBank # L22060.1).
Matches required a minimum percent identity of 90%, and a Smith-Waterman score larger than 18.
Read pairs where at least one of the pair met mapping criteria were discarded. This procedure removed 0.44% of the reads. We also identified and masked adapter sequences based on BLAST searches of Illumina multiplexed, paired-end mRNA-Seq adapters against all reads, requiring an e-value less than 100 for a match.
0.007% of reads were masked using this procedure, suggesting very low adapter contamination.
Illumina libraries often suffer from lower k-mer diversity in the first 12-15 bp of each read, suggesting that the random priming step of Illumina mRNA-Seq library
preparation is not entirely random. As a result, the first ~15 bp of an Illumina library usually has lower k-mer diversity. Preliminary analyses showed that the assembly N50 was lower in libraries where we did not remove the first 15 bp (data not shown). Because of this, we trimmed the first 15 bp of each read in our libraries.
Regions with a phred score equivalent of less than 20, as well as masked contaminant sequence, were trimmed using SolexaQA DynamicTrim
[6]
. All sequences shorter than 25 bp were removed using LengthSort from the same package. Overall,
9.44% of all reads were removed in our quality control steps.
Assembly
Our assembly scheme loosely followed the “STM” approach introduced by Surget-Groba and Montoya-Burgos
[7]
. This approach first assembles the reads de novo into contigs, and then scaffolds these contigs onto reference protein and DNA sequence sets. Contigs that overlap on scaffolds are then re-assembled. To improve the gene content and contig length of the assembly, we added contigs derived from 454 GS-FLX sequences of Ae. albopictus oocyte mRNA
[8]
. Below we describe each step of the assembly procedure in turn.
Initial de novo assembly . We used Velvet
[9]
followed by Oases
[10]
to assemble cleaned reads by development stage and light treatment Figure S1, Step 1). Initial exploratory assemblies showed that a lower percentage of reads were used in the final assembly when more data was included, and that performing multiple, smaller assemblies resulted in higher read usage. Reads that Oases did not include in the initial four assembly groups were combined for an additional assembly. Assemblies were performed
from kmers 19 to 59 with 10 kmer-intervals at a coverage cutoff of 10. Contig redundancy of near-identical contigs resulting from separate kmer assemblies was eliminated using CD-HIT-EST
[11]
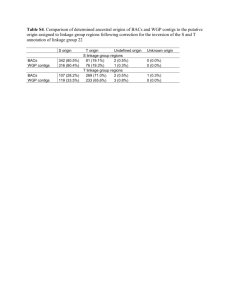
, which groups short sequences with longer ones over a user-defined identity threshold. We required 99% identity, otherwise using the default parameters. This procedure reduced the total number of contigs from 732,706 to 293,530
(Table S1).
Merged reference assembly.
We used two complementary sets of reference sequences to scaffold, merge and annotate de novo assembled contigs: a Dipteran protein set derived from OrthoDB
[12]
, and the Ae. aegypti genome sequence
[13]
. We downloaded the
Diptera-specific ortholog set from OrthoDB v.4, and retained all Ae. aegypti proteins from each ortholog group. If an ortholog group did not have an Ae. aegypti representative, then a protein from the series of closest relatives to Ae. albopictus was chosen, in the order of Culex pipiens, Anopheles gambiae, and Drosophila melanogaster .
This resulted in a reference set of 21,066 protein sequences. The objective of this approach was to maximize the diversity of proteins in the reference set by including a wide taxonomic range, while simultaneously reducing the redundancy of the proteins in the set by only including proteins from one species per ortholog group.
We aligned all contigs derived from the initial de novo assembly, as well as contigs from a separate assembly of 454 transcripts from Ae. albopictus oocyte tissue
[8]
, to the masked Dipteran protein set using the program fastx
[14]
. We required an e-value of
1e-6 for an alignment to be scored, and assigned a contig to the reference protein with the lowest e-value. Contigs assigned to the same reference protein were grouped and re-assembled in cap3
[15]
, requiring 95% identity of overlapping regions. Annotations of
the re-assembled contigs were updated by aligning them to the reference protein set using the same fastx parameters; only contigs with > 70% identity to their best match were retained for further analysis. We identified potential chimeric contigs by searching for additional alignments to a given contig that were outside the primary alignment, and within 80% of the primary alignment's percent identity; contigs that fit these criteria were discarded. Overall, 46.7% of all contigs matched the Dipertan protein set, 90.9% of these were re-assembled, and 7.5% of the re-assembled contig set were chimeric. 33,532 contigs were generated in this procedure, from an initial 169,495, demonstrating the ability of the approach to reduce contig redundancy (Table S1).
Remaining contigs that did not meet the fastx criteria were aligned to Ae. aegypti supercontigs using exonerate
[16]
(parameters were est2genome --softmaskquery --bestn
1 --dnahspdropoff 0). To reduce computational time, contigs were matched to Ae. aegypti supercontigs using megablast (e-value <1e-6), and subsequent exonerate alignments were only performed on the matched supercontig. To reduce the number of spurious annotations, only alignments with over 72.76% identity (the 95 th percentile of all alignments) were used. Contigs with overlapping alignments (>1 bp) were then reassembled in cap3 as above, and re-aligned to the Ae. aegypti supercontigs for annotation.
As above, only contigs with >70% identity to the reference were retained. We grouped the re-assembled contigs into two categories: contigs that aligned within gene models or potential UTRs, and contigs that aligned outside of gene models or potential UTRs. First, contigs that aligned within gene models or potential UTRs were annotated by their corresponding gene model. We defined matches to potential UTRs as contigs that aligned within 1kb of a gene model and over 90% of the contig’s length. Second, contigs that
aligned outside of gene models and potential UTRs were only annotated by their coordinates on the supercontig. Because Ae. aegypti can have very large introns
[13]
, we used the default maximum intron size (200,000 bp). As a likely result of this, 12.4% of contigs had alignments that spanned multiple gene models. These contigs were considered chimeric mis-assemblies and were discarded. The generation of contigs via exonerate alignments and re-assemblies added an additional 17,194 annotated contigs to the assembly, from an initial 71,435 megablast alignments. The final, annotated assembly was comprised of 50,725 contigs (Table S1). The mean/median contig length increased from 1,033.7/555 to 2,340/1,818 after the re-assembly.
The goal of this alignment procedure was to reduce the possibility of spurious annotations. As a result, many contigs were not annotated; however, the conservative set of annotated contigs used in subsequent analyses can be interpreted with high confidence.
Blastx and Blastn analyses of the un-annotated contigs against the SwissProt database, three Wolbachia strains, a previous assembly of an Ae. albopictus oocyte transcriptome assembly
[8]
, and RepBase were able to match over half of the un-annotated contigs. The majority of these matches occurred between the two Ae. albopictus transcriptomes. This suggests that some sequences of the Ae. albopictus transcriptome expressed at both the oocyte and embryo stages are too diverged from other reference organisms to assign homology, despite the relatively close evolutionary relationships between Ae. albopictus and these references. Differential expression analysis of these transcripts will be performed once the Ae. albopictus genome sequence is available.
The final, annotated assembly identified 12,345 gene models, with some overlap between contigs from each reference assembly. Overall percent identities between
contigs and their references were high, as were the proportion of the contig and reference in the alignment, (Figures S2-3). In the genomic alignments that matched gene models, the proportion of the reference gene model covered by genomic alignments was on average low, suggesting that many alignments in this analysis could occur in unannotated areas of the Ae. aegypti genome sequence (Figure S3) .
The reference-based merging procedure effectively doubled the annotated average contig length from 1031.55 in the de novo assembly to 2015.93 in the final annotated assembly (Table S1, Figure
S2).
Gene expression quantification, analysis and validation
De novo transcriptome assemblies often generate multiple contigs per unigene as a result of isoform or allele assembly, or due to incomplete transcript coverage. This redundancy can complicate gene expression quantification from read mapping, because reads may map to multiple contigs. Therefore, we quantified expression for each unigene using
RSEM
[17]
, which generates read counts on a per-gene basis from contig assemblies.
RSEM uses bowtie (version 0.12.7)
[18]
to map reads to contigs, and uses the output to calculate expression by unigene, rather than single contigs. We used the program’s default parameters. Paired-end and single-end reads, both of which were contained in our dataset after read-cleaning, cannot be combined in an analysis. Therefore, we only mapped paired-end reads; we performed trial, single-end mappings using the first read of each read pair, and “singleton” reads lacking a read-pair after read cleaning, and obtained similar results (data not shown).
The resulting read counts were processed in R ( http://www.r-project.org/ ). While all annotated contigs were used in expression quantification, we removed contigs that aligned to un-annotated regions of the Ae. aegypti genome from this stage onward,
because 1) these contigs tended to have very low expression estimates that would likely not lead to differential expression detection, and 2) our downstream analyses required functional annotation. We also removed all gene models with fewer than one count per million reads in three or fewer libraries, as these genes likely have too low expression to be detected as significant, which would contribute to loss of power in the analysis (e.g.
Robinson et al. 2012). This step removed 895 genes from the analysis (the average total read-count from the removed genes was 19.2; maximum, 82). The data were then TMMnormalized in edger
[19]
, which accounts for both library size and expression bias.
Normalized read counts are available in Table S1.
To independently verify the gene expression results, we compared results from the RNA-Seq data with relative mRNA abundance data from Ae. albopictus lipid metabolism genes in Reynolds et al .
[1]
. Tissue generation and RNA extraction was performed under the same experimental conditions and in the same laboratory for both studies, where the Reynolds et al . study used material from laboratory F
10
-F
11
generations of the same Ae. albopictus strain. Gene acs ( Ae. aegypti homolog: AAEL017299) was not identified in the RNA-Seq data, which is consistent with the low mRNA abundance detected from qRT-PCR
[1] . Pearson’s correlation coefficient was calculated from the gene expression data, averaged across replicates, between qRT-PCR and RNA-Seq estimates for each gene, photoperiod treatment and development stage. Gene expression estimates from RNA-Seq and qRT-PCR were highly congruent (overall correlation,
Pearson’s r = 0.92; Figure S6).
Transcript length heterogeneity in RNA-Seq datasets can bias enrichment analyses: longer transcripts can generate higher read counts, which are more likely to be
detected as DE. Thus, a category comprised of longer gene models is more likely to contain differentially expressed genes. This potential discrepancy among categories can lead to bias in the selection of enriched categories. We chose the following approach to ascertain whether transcript length bias was prevalent in our dataset. The “GO-Seq” program [20] performs both the “Wallenius” method, which corrects enrichment analyses for transcript length bias, as well as the “Hypergeometric” method, which does not. We reasoned that if enriched categories differ between these two methods, then significant length bias exists; if there is no substantial difference, then alternative enrichment analyses that do not correct for transcript length, such as the DAVID Bioinformatics
Resource [21,22], are feasible alternatives. We performed the GO-Seq analysis with GO-
FAT and Kegg categories provided from the DAVID bioinformatics database. Transcript lengths were calculated as the median contig length for each unigene in our dataset.
Identical to the DAVID analysis described in the main text of this article, D. melanogaster orthologs for each gene model were used (one-to-one and apparent one-toone, retrieved as Flybase gene IDs from BioMart [12,23]
)
. Almost all of the same categories were enriched for both transcript length-corrected and uncorrected methods, as determined by a Benjamini-Hochberg corrected p-value < 0.05, at either 3d or 6d pov
(results not shown). There was only one exception, the category “DNA metabolic process”, that was significantly enriched using the uncorrected method at 3d pov
(p=0.0466), but not in the bias-corrected method (p=0.0534). However, this category did not add any new information, since it is the parent category for two other enriched categories (“DNA replication” and “DNA replication initiation”). Based on these
analyses, we conclude that there is no meaningful transcript length bias in our dataset, and that analyses with the DAVID Bioinformatics resource are valid.
Literature Cited
[1] Reynolds, J. A., Poelchau, M. F., Rahman, Z., Armbruster, P. A. & Denlinger, D. L.
2012 Transcript profiling reveals mechanisms for lipid conservation during diapause in the mosquito, Aedes albopictus. J. Insect Physiol. 58 , 966-973. (DOI http://dx.doi.org/10.1016/j.bbr.2011.03.031).
[2] Armbruster, P. A. & Hutchinson, R. A. 2002 Pupal mass and wing length as indicators of fecundity in Aedes albopictus and Aedes geniculatus (Diptera : Culicidae) .
J. Med. Entomol. 39 , 699-704.
[3] Armbruster, P. A. & Conn, J. E. 2006 Geographic variation of larval growth in North
American Aedes albopictus (Diptera: Culicidae) . Ann. Entomol. Soc. Am. 99 , 1234-1243.
[4] Mori, A., Oda, T. & Wada, Y. 1981 Studies on the egg diapause and overwintering of
Aedes albopictus in Nagasaki . Tropical Medicine. 23 , 79-90.
[5] Ning, Z. M., Cox, A. J. & Mullikin, J. C. 2001 SSAHA: A fast search method for large DNA databases . Genome Res.
, 1725-1729.
[6] Cox, M. P., Peterson, D. A. & Biggs, P. J. 2010 SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data . BMC Bioinformatics. 11 , 485.
[7] Surget-Groba, Y. & Montoya-Burgos, J. 2010 Optimization of de novo transcriptome assembly from next-generation sequencing data . Genome Res. 20 , 1432-1440. (DOI
10.1101/gr.103846.109).
[8] Poelchau, M. F., Reynolds, J. A., Denlinger, D. L., Elsik, C. G. & Armbruster, P. A.
2011 A de novo transcriptome of the Asian tiger mosquito, Aedes albopictus , to identify candidate transcripts for diapause preparation . BMC Genomics. 12 (DOI 10.1186/1471-
2164-12-619).
[9] Zerbino, D. R. & Birney, E. 2008 Velvet: Algorithms for de novo short read assembly using de Bruijn graphs . Genome Res. 18 , 821-829. (DOI
10.1101/gr.074492.107|10.1101/gr.074492.107).
[10] Schulz, M. H., Zerbino, D. R., Vingron, M. & Birney, E. 2012 Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels . Bioinformatics.
28 , 1086-1092. (DOI doi: 10.1093/bioinformatics/bts094).
[11] Li, W. Z. & Godzik, A. 2006 Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences . Bioinformatics. 22 , 1658-1659. (DOI
10.1093/bioinformatics/btl158|10.1093/bioinformatics/btl158).
[12] Waterhouse, R. M., Zdobnov, E. M., Tegenfeldt, F., Li, J. & Kriventseva, E. V. 2011
OrthoDB: the hierarchical catalog of eukaryotic orthologs in 2011 . Nucleic Acids Res. 39 ,
D283-D288. (DOI 10.1093/nar/gkq930|10.1093/nar/gkq930).
[13] Nene, V., Wortman, J. R., Lawson, D., Haas, B., Kodira, C., Tu, Z., Loftus, B., Xi,
Z., Megy, K., Grabherr, M., et al . 2007 Genome Sequence of Aedes aegypti , a Major
Arbovirus Vector . Science. 316 , 1718-1723. (DOI 10.1126/science.1138878).
[14] Pearson, W. R., Wood, T., Zhang, Z. & Miller, W. 1997 Comparison of DNA sequences with protein sequences . Genomics. 46 , 24-36.
[15] Huang, X. & Madan, A. 1999 CAP3: A DNA sequence assembly program . Genome
Res. 9 , 868-877.
[16] Slater, G. & Birney, E. 2005 Automated generation of heuristics for biological sequence comparison . BMC Bioinformatics. 6 , 31.
[17] Li, B. & Dewey, C. 2011 RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome . BMC Bioinformatics. 12 , 323.
[18] Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. 2009 Ultrafast and memoryefficient alignment of short DNA sequences to the human genome . Genome Biol. 10 ,
R25.
[19] Robinson, M. D., McCarthy, D. J. & Smyth, G. K. 2010 edgeR: a Bioconductor package for differential expression analysis of digital gene expression data .
Bioinformatics. 26 , 139-140.
[20] Young, M., Wakefield, M., Smyth, G. & Oshlack, A. 2010 Gene ontology analysis for RNA-seq: accounting for selection bias . Genome Biology. 11 , R14.
[21] Huang, D. W., Sherman, B. T. & Lempicki, R. A. 2009 Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists . Nucleic
Acids Res. 37 , 1-13. (DOI 10.1093/nar/gkn923).
[22] Huang, D. W., Sherman, B. T. & Lempicki, R. A. 2009 Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources . Nature Protocols. 4 ,
44-57. (DOI 10.1038/nprot.2008.211).
[23] Haider, S., Ballester, B., Smedley, D., Zhang, J. J., Rice, P. & Kasprzyk, A. 2009
BioMart Central Portal-unified access to biological data . Nucleic Acids Res. 37 , W23-
W27. (DOI 10.1093/nar/gkp265).
All tables of the electronic supplementary material are given in a separate Excel file due to their size.
Figure S1. Flowchart demonstrating transcriptome assembly steps. The first stage constructed a de novo assembly using the multi-k method. The second stage reduced the redundancy and increased contig length by scaffolding contigs against references sequences and re-assembling them (c.f. Surget-Groba and Montoya-Burgos 2010).
Figure S2. Box plots of contig length distributions from different stages of the assembly.
Note the log
10
scale of the y-axis.
Figure S3 . %Identity (a), proportion of contig in the alignment (b), and proportion of reference in the alignment (c) of re-assembled contigs resulting from alignments to the protein sequence and genomic references.
Figure S4. Scatter-plot of relative mRNA abundance estimates and TMM-normalized read-counts. Relative mRNA abundance values were derived from qRT-PCR experiments, and TMM-normalized read-counts from RNA-Seq data, from 20 candidate genes analyzed in Reynolds et al. [16]. Each point represents an individual gene’s expression at either 3d or 6d, under D or ND photoperiod conditions.
Figure S5 . Example embryo images. Pictures of 3d ND embryos were taken from laboratory generation F11; all other pictures were taken from laboratory generation F10, which was sequenced. Calculations of the percentage of embryos in each developmental
stage were performed on full sets of embryos within the F10 laboratory generation (see
Additional File 1). Embryos that were scored as deviations from the main morphological range are marked by arrows. Panel a), D embryos at 3d post-oviposition, laboratory generation F10; b), ND embryos at 3d post-oviposition, laboratory generation F11; c), D embryos at 6d post-oviposition, laboratory generation F10; d), ND embryos at 6d postoviposition, laboratory generation F10.
Figure S6.
Hierarchical clustering dendrogram of global Ae. albopictus expression patterns using the default settings of the function pvclust in R (Suzuki and Shimodaira
2006). Average clustering of complete gene expression profiles was used to generate the dendrogram. Correlation-based dissimilarity between gene expression profiles is listed on the y-axis. Tips of the dendrogram represent each of the 12 libraries sequenced. All branches have approximately unbiased bootstrap values > 0.84.
Figure S7 . Heatmap of Ae. albopictus and D. melanogaster 1:1 orthologs for the Gene
Ontology category “cell cycle process”. Expression values are depicted as z-standardized scores, where blue represents low expression, and yellow, high. Expression values from all twelve Ae. albopictus libraries are shown. D. melanogaster expression is shown from
8 hours – 22 hours post-oviposition.