tpj12967-sup-0024-Legends
advertisement

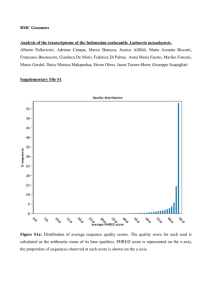
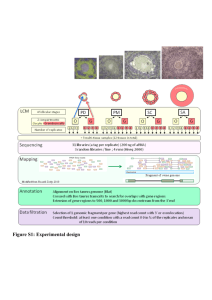
Supporting Information Legends Figure S1: Screenshots showing the 6 reading frames of sequences of the Multi-kmer contig assembly with the software RESearch (Amersham). Start codons are shown in blue, stop codons in red and Open Reading Frames between Start and Stop codons are in green. a) Example of sequence with no obvious reading frame; b) Example of a trans-self chimera sequence, probably due to an assembly error; c) Example of a sequence containing 2 transcripts assembled by their 3’ UTR; d) Different isoforms of a galactinol synthase gene found in the assemblies. 1: Transcript potentially coding for a 334 amino acid protein; 2: Alternative splice transcript potentially coding for a 327 amino acid protein; 3: Transcript with the retention of 2 introns. Figure S2: Distribution of contig lengths in three different assemblies of the pea RNA-seq reads. All contigs from PsCameor_LowCopy, PsCamTri1E_LC and PsCamTri2E_LC were distributed according to their length. The number of contigs is expressed as a percentage of the total number of contigs in each assembly. Figure S3: Distribution of Gene Ontologies of the pea Unigene contigs. a, b: GO for Biological processes, c,d: GO for Molecular function, a,c: PsCam_HichCopy Unigene contigs, b,d: PsCam_LowCopy Unigene contigs. Figure S4. Gene expression specificity across pea tissues. All pea transcripts (red line), unannotated transcripts (green line) and transcription factor transcripts (blue line) were classified into four groups according to their tissue specificity: not differentially expressed (fold-change<3 between the tissues showing the highest and the second highest expression levels), preferentially (fold changes >= 3 and <10), very preferentially (fold changes >=10- and <100), specifically (fold changes >=100 and <1000) and very specifically expressed in one tissue (fold changes >=1000). Figure S5: Distribution of the putative molecular functions of specifically expressed pea contigs. Genes specifically and very specifically expressed in Apical Nodes, Flowers, Nodules, Roots and Seeds are presented. Genes with no assigned molecular function were not considered. Figure S6. Distribution of pea transcription factors expressed at least specifically in one tissue, based on their family membership. The sub-pies highlight the distribution of specific transcription factor gene families in the different tissues, based on the specificity of their expression. Figure S7: K-means clustering of RPKMnorm expression profiles of PsCam_LowCopy Unigene contigs. The y-axis expresses the mean level of expression in each cluster. The x-axis indicates the different samples in the following order: RootSys_A_HN, RootSys_A_LN, Root_B_LN, Root_F_LN, Nodule_A_LN, Nodule_B_LN, Nodule_G_LN, Shoot_A_HN, Shoot_A_LN, Leaf_B_LN, LowerLeaf_C_LN, UpperLeaf_C_LN, Tendril_BC_LN, Stem_BC_LN, Peduncle_C_LN, ApicNode_B_LN, Flower_B_LN, Pods_C_LN, Seeds_12dap, Seed_5 dai. Error bars represent Mean Standard Error. Figure S8: TopGO analysis of differentially expressed genes between nodules and shoots. Gene ontology (GO) for a. Biological Processes (BP), b. Molecular Function (MF) and c. Cellular Component (CC) are depicted. Figure S9: TopGO analysis of differentially expressed genes between roots and nodules. Gene ontology (GO) for a. Biological Processes (BP), b. Molecular Function (MF) and c. Cellular Component (CC) are depicted. Figure S10: Distribution of differences in predicted peptide lengths between pea and M. truncatula (a) or G. max (b) orthologous genes. Table S1: Analysis of splicing variants retrieved from the different RNA-seq assembly for 8 pea known genes. TableS2: Distribution of contigs showing low, not preferential, preferential, very preferential, specific, very specific tissue expression according to differential expression and to tissue localization of maximum RPKMnorm expression. TableS3: List of contigs putatively encoding transcription factors and showing very preferentially and specifically expressed in a tissue expression. Expression and annotation information are provided (TF Family, IPR and GO annotation, RPKMnorm max and RPKMnorm in the 20 libraries). Table S4: Contig distribution among K-means clustering groups according to tissue localization of maximum RPKMnorm expression. Table S5: Contig distribution among K-means clustering groups according to differential expression classes as defined in Table 6. TableS6: List of contigs significantly and very preferentially, specifically or very specifically expressed in nodules. This table summarizes all the information from the differential expression (DE class, Specificity, RPKM max, K-group, DEseq results between Nod_Shoot, Root_Shoot, Nod_Root) and the annotation analyses (InterProScan, GO, Mapman BIN, best homologues in Medicago truncatula, Glycine max, Cajanus cajan, Arabidopsis thaliana). Table S7: Number of transcript clusters generated for the PsCam_LowCopy and PsCam_HighCopy Unigene sets using OrthoMCL (Li et al. 2003). Each cluster consists of homology gene families from pea and M. truncatula. It comprises ortholog or recent paralog transcript from both taxa. Singletons correspond to transcripts that did not cluster in any homology gene family. Single Copy families correspond to clusters with only one member in each taxa. Table S8: List of pea best homologues of major determinants of nodulation in M. truncatula and L. japonicus. This table summarizes for all genes, the pathway, the stage of expression, the annotation in pea, M. truncatula, L. japonicus, the specificity class, the organ showing maximum RPKMnorm, the Differential Expression group, the levels of expression in the 20 tissues, and the annotations by InterProScan and by homology to M. truncatula, G . max, C. cajan, A. thaliana. Table S9: List of tissues sampled for RNA-seq, according to the experiment, stage of harvest, and growing condition. Table S10: Development and growth characteristics of 4 plants harvested at Stages A (06/04/2010), B (19/04/2010) and C (07/05/2010). Table S11: RNA-seq read specifications. Table S12: Contig assembly characteristics after Velvet-Oases steps, using the multi_kmer strategy. Table S13: Primer sequences used for the validation of in silico expression levels by qPCR expression level quantification.