Chemical Transformations
advertisement

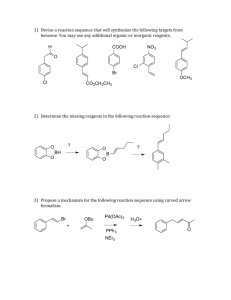
Chemical Transformation Problem Statements Problem 1: Asymmetric Hydrogenation Asymmetric hydrogenation of C=C and C=Het is a powerful strategy in synthesis. Whilst the asymmetric hydrogenation of isolated acyclic C=Het bonds is largely a solved problem the reduction of simple C=C and unsaturated heterocycles remains a key challenge.1 In the case of C=C the asymmetric variant is reliant on the presence of key functional groups for success. The use of chiral phosphine ligands co-ordinated to rhodium to reduce unsaturated aminoacrylates is well precedented. Although the scope of the reaction can be extended using bulkier ligands or amino-phosphine based systems. The asymmetric reduction of heterocycles often requires the use of high pressure to achieve successful reaction and ee’s are variable. Expanding the range of substrates and methodologies in the asymmetric reduction of C=C and heterocycles would provide a key tool to the synthetic chemist. Exemplification of Problem HO2C NH2 H N R N H H N R Y N R3 R2 R1 The enantiomerically pure saturated heterocycles shown above are found in GSK development compounds but represent a real challenge for synthesis, requiring multiple steps and/or classical resolution. These fragments (and many more) could be prepared by asymmetric hydrogenation of the corresponding unsaturated heterocycles, thus avoiding the need for stoichiometric resolving agents and offering yields over the inherent 50% limit for resolution. Expected Output of Research The identification and development of suitable catalyst systems which would enable the scalable reduction of unsaturated heterocycles and simple olefins. Renewable solvents should be considered as media for these reactions, and they should demonstrate improved ‘green’ metrics versus the transformation they are replacing. A view towards broader application and industrialization should also be provided. 1 X. Zhang et al., Acc Chem Res., 2007, 40, 1278 and H. Shimizu et al., Acc. Chem. Res., 2007, 40, 1385. Problem 2: Remote C-H oxidative transformations The seminal book written by Corey and Cheng twenty years ago taught us how to disconnect a complex molecule to simpler starting materials using retrosynthetic analysis. Although remote C-H functionalisation was mentioned in this monograph it was an underdeveloped area. Catalytic C-H functionalisation is now revolutionizing the way of thinking in organic synthesis with a growing number of reviews in the literature.2 Exemplification of Problem Manganese and iron–based catalysts have been used with both molecular oxygen and hydrogen peroxide to oxidize a range of systems, including remote C-H bonds.3 However, the activity of these systems is difficult to tune with significant problems of over-oxidation and chemoselectivity. Further work is needed to provide better predictive models for reactivity and chemoselectivity.4 In addition, new methodologies are needed to expand the scope and utility of the area. Catalytic C-H amination via insertion of a metal nitrene species is another exciting area of research. Intramolecular and intermolecular variants are exemplified by the work of Breslow5 and Du Bois.6 Expanding the scope and utility of catalytic methodologies to synthesise amines is a key part of GSKs sustainability strategy. Expected Output of Research The identification and development of suitable catalysts which would enable the scalable and predictable oxidative transformation of remote C-H bonds. The functionalisation of C-H bonds to introduce halogen, oxygen and nitrogen can be considered as a formal oxidation process. Renewable solvents should be considered as media for these reactions, and they should demonstrate improved ‘green’ metrics versus the transformation they are replacing. A view towards broader application and industrialization should also be provided. If oxygen or hydrogen peroxide is used, safe use in a Pharmaceutical environment should be considered. “Selelective functionalisation of C-H bonds” in Chem Rev., 2010, 575-1211 B. L. Feringa et al., J. Am. Chem. Soc., 2005, 127, 7990 and C. White et al., Science, 2007, 318, 783 4 C. White et al., Science, 2010, 327, 566. 5 R. Breslow et al., J. Am. Chem. Soc., 1983, 105, 6728. 6 J. Du Bois et al., J. Am. Chem. Soc., 2001, 123, 6935 and J. Am. Chem. Soc., 2004, 126, 15378. 2 3 Problem 3: Green Oxidations (in Air, e.g.) Oxidations, as a broad class of reactions, can be some of the most powerful reactions used in organic chemistry. Unfortunately, they are often plagued by high molecular weight oxidants and requirements for large excesses of toxic metals which lead to negative environmental impacts if used on scale. Recent work using sub-stoichiometric amounts of metal catalysts in concert with environmentally friendlier oxidizing reagents has led the way towards greener approaches to oxidations. One such approach centers around the use of catalysts (both metal and non-metal) in the presence of molecular oxygen or air.17 Several “green” advantages to this approach include, the use of a cheap and atom economical oxidant, the use of catalytic amounts of metals or co-oxidants and minimization of toxic waste streams (filtration and recycling of metal catalyst). An important consideration with the use of oxygen as a stoichiometric, terminal oxidant is flammability. Use in water or with nitrogen dilution should be strongly considered. Alternatively, engineering solutions may be possible. A range of approaches have emerged including the use of palladium catalysis, hypervalent iodine (HVI) compounds, Pt/Bi on carbon, etc. Exemplification of Problem8,9 R2 R1 Pd cat., O2 HOAc OAc R2 R1 K2CO3 MeOH OAc OH R1 R2 OH O OH PhI(OAc)2, TEMPO, KNO2 O2, 80 oC R R Expected Output of Research 7 The identification and development of suitable catalyst systems which enable scaleable oxidations. Use of molecular oxygen as the terminal oxidant (or some comparable, low MW, safe, low impact oxidant) in non-halogenated solvents. Flammability of reactions systems should be considered. Thus oxidations in water are preferred or oxidations with the use of dilute oxygen (dilution with nitrogen, e.g.) or ones which incorporate an engineering solution. A view towards broader application and industrialization should also be provided. Mallat, T.; Baiker, A. Chem Rev. 2004, 104, 3037. Wang, A.; Jiang, H.; Chen, H. J. Am Chem. Soc. 2009, 131, 3846. 9 Herrerias, C.; Zhang, T.; Li, C-J. Tetrahedron Lett. 2006, 47, 13. 8 Problem-4: Amide Bond Reduction Without Hydride Reagents Amide reduction can be a preferred approach to amine synthesis as a way of eliminating the need to make or handle alkylating agents associated with N-alkylation strategies. Common methods for reduction of amide bonds however, involve the use of metal hydrides (LiAlH4, DIBAL, RedAl, etc.), diborane, Et3SiH or polymethylhydrosiloxane (PMHS). Many of these methods can lead to product isolation issues (slow filtrations, product occlusion, etc) and waste disposal problems (e.g. aluminum waste). Exemplification of Problem One of the steps in the synthesis of Paroxetine involves the reduction of a piperidone to the corresponding piperidine using LiAlH4.10 While LiAlH4 has a favorable hydride density,1 aluminum waste disposal (aluminum hydroxide salts) is an issue for highvolume products. Ar Ar CO2Me N O R OH N R Hydrogen gas is the ideal reducing agent as the only by-product is water. While there has been progress in area of metal-catalyzed reduction of amides using hydrogen gas,11 current methods require high temperatures and pressures. Pressures of these magnitudes are not routinely available in a typical pharmaceutical manufacturing plant. Expected Output of Research Identification and development of suitable catalysts which would enable direct amide bond reduction with the use of hydrogen gas or another atom-economical reducing agent under relatively mild temperatures and pressures. Renewable solvents should be considered as media for these reactions, and they should demonstrate improved ‘green’ metrics versus the transformation they are replacing. A view towards broader application and industrialization should also be provided. If hydrogen is used, low pressures are preferred to minimize installation costs. Electrochemical methods may be considered. 10 Yu, M.; Lantos, I.; Peng, Z-Q.; Yu, J.; and Cacchio, T. Tetrahedron Lett. 2000, 5647. (a) Magro, A.; Eastham, G.; Cole-Hamilton, D. Chem. Commun. 2007, 3154. (b) Hirosawa, C.; Wakasa, N.; Fuchikami, T. Tetrahedron Lett. 1996, 6749. 11 Problem-5: Greener Mitsunobu Alternatives The Mitsunobu reaction is a powerful way to substitute alcohols with a range of competent nucleophiles and in the case of secondary alcohols, with high stereospecificity. The reaction has also been applied intramolecularly in the synthesis of lactones, lactams and macrocycles. For a comprehensive and contemporary review of the Mitsunobu reaction, see Swamy, et al.12. The primary drawback of this reaction from an environmental perspective is the stiochometric requirement for azodicarboxylates and trialkyl- or triarylphosphines which negatively impact overall atom economy.1 Further, purification issues often arise from stoichiometric by-products that are derived from the use of these reagents. Exemplification of Problem The overall conversion in the Mitsunobu reaction is a substitution reaction accompanied by a redox reaction (mediated by trialkyl- or triarylphosphines and azodicarboxylates). The power of the Mitsunobu reaction lies with the sterochemical fidelity with which secondary alcohols can be substituted. Further benefits include predictability, scope of competent nucleophiles and moderate operating temperature. A number of approaches11 have been taken to improve isolation issues including modifications to azocarboxylates and phosphines which alter solubility (e.g. acid/base/water). Further, derivatives of these reagents have been attached to solid supports. While these approaches may facilitate isolation on small scale, they still present significant drawbacks (particularly solid supported reagents) in terms of environmental impact and atom economy. Step improvements have also been made with improving atom economy be combination of redox partners within a single reagent. 13 Expected Output of Research Ideally catalytic methods would be developed that achieve the overall net transformation observed with the classical Mitsunobu reaction. The new method(s) would eliminate the need for phosphines, azodicarbolxylates or other stoichiometric reagents that lead to isolation issues, poor atom economy or poor Mass Intensity. Renewable solvents should be considered as media for these reactions, and they should demonstrate improved ‘green’ metrics versus the transformation they are replacing. A view towards broader application and industrialization should also be provided. 12 13 Swamy, K.; Kumar, N.; Balaraman, E.; Kumar, K. Chem. Rev. 2009, 109, 2551. Tsunoda, T.; Nagino, C.; Oguri, M.; Ito, S. Tetrahedron Lett. 1996, 37, 2459. Problem-6: Greener OH Activation Methods Due to the poor leaving group ability of hydroxide ion, methods for alcohol displacement typically require pre-activation in the form of halide or sulfonate ester derivatives or alternatively the use of, in some cases, stoichiometric quantites of Bronstead or Lewis acids (with the net displacement of water). Thus an atom-efficient method for the direct displacement of alcohols with a range of nucleophiles is desirable. Exemplification of Problem Alkyl, allylic or benzylic halides and sulfonate esters often pose significant handling and purification issues due, in part, to their potential genotoxicity. Furthermore, sulfonate esters are not atom economical. Alternatively, strong Brønsted or Lewis acids have been used for alcohol displacements. These methods are, at times, limited by substrate sensitivities, functional group incompatibilities or selectivity issues. The use of stoichiometric Lewis acids naturally leads to significant waste. It is well known that allylic acetates have been used extensively in Pd-catalyzed substitution reactions with a variety of nucleophiles. More recently, allyic carbonates have been used, for example, in iridium-catalyzed amination reactions.14 Even more promising is the use of iridium15, palladium16 or platinum17 catalysis for the direct conversion of allylic alcohols. Expected Output of Research Ideally, methods could be developed which involve catalytic activation of alcohols which would enable displacement by a range of competent nucleophiles. In the case of secondary alcohol substrates, stereochemical integrity (either retention or inversion) would be preserved. The additional challenge would be expansion of scope to include a broad range of alcohol substrates. Renewable solvents should be considered as media for these reactions, and they should demonstrate improved ‘green’ metrics versus the transformation they are replacing. A view towards broader application and industrialization should also be provided. 14 Pouy, M.; Stanley, L.; Hartwig, J. J. Am. Chem. Soc. 2009, 131, 11312. Defieber, C.; Ariger, M. Moriel, P.; Carreira, E. Angew. Chem. Int. Ed. 2007, 46, 3139. 16 Nishikata, T.; Lipshutz, B. Organic Lett. 2009, 11, 2377. 17 Ohshima, T.; Miyamoto, Y.; Ipposhi, J.; Nakahara, Y.; Utsunomiya, M.; Mashima, K. J. Am. Chem. Soc. 2009, 131, 14317. 15 Problem 7: An efficient synthesis of oligonucleotides Synthetic oligonucleotides have become a new class of therapeutic agents in recent years. Currently oligonucleotides are synthesized by the phosphoramidite-based solid phase synthesis. Although this process provides high quality oligonucleotides at quantities required for clinical development, it requires excess reagents and extremely large volume of solvents, hence is not considered to be sustainable, and not suitable for the large scale commercial production. Exemplification of Problem The current phosphoramidite method had become the “method of choice” to synthesize oligonucleotide since late 1990’s as a group from ISIS pharmaceuticals established the automated process. Since then, numerous modifications/improvements have been made to make this process more efficient, as a result, it becomes the standard method for the oligonucleotide synthesis (Scheme 1).18 Scheme 1. Phosphoramidite based solid phase oligonucleotide synthesis 18 Capaldi, Daniel C.; Scozzari, Anthony N., Antisense Drug Technology (2nd Edition) (2008), 401-434 Besides the phosphoramidite method, on the other hand, there are numerous reports utilizing different modes of coupling methods, such as H-phosphonate approach, phosphate trimester approach, and phosphotriester approach. Because the majority of the focus has been devoted to phosphoramidite chemistry for the past decade, these alternative approaches are still underdeveloped.19 The phase of synthetic process is another consideration. The solid phase synthesis is currently employed in all practical oligonucleotide syntheses. While the solid phase synthesis has clear advantages over solution phase syntheses, such as easy removal of excess reagents, it normally requires large excess reagents/solvents to achieve high chemical conversion. Other media have been explored (ionic liquid, solution phase, PEG, Fluorous), but all approaches are still considered to be primitive.20,21 Expected Output of Research Development of a synthetic process which uses significantly less volume of solvents than the current solid phase synthesis. Demonstration of the process which enables production of 15-20 mer oligonucleotides with comparable quality to the current state of the solid phase synthesis. Ideally, the new process would be capable of scaling up to >10 kg scale. 19 C. B. Reese, Org. Biomol. Chem., 2005, 3, 3851 Bonora G. M.; Rossin R.; Zaramella S.; Cole D. L.; Eleuteri A.; Ravikumar V. T., Org. Process Res. Dev., 2000, 4, 225 21 Donga R. A.; Hassler M.; Chan T-H.; Damha M. J., Nucleosides, Nucleotides nucleic Acids, 2007, 26, 1287 20