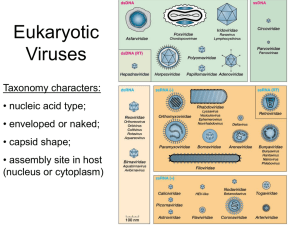
Our_Abstractbook_***** – SPINE 2 - SPINE2
advertisement

VIZIER / SPINE2 Workshop on Structural Virology 14th - 16th July, 2008 Vienna Organizing committee Martino Bolognesi Dept. of Biomolecular Sciences & Biotechnology University of Milano Via Celoria, 26 20131 Milano – Italy martino.bolognesi@unimi.it Kristina Djinovic Department for Biomolecular Structural Chemistry Max F. Perutz Laboratories University of Vienna, Campus Vienna Biocenter 5 A-1030 Vienna, Austria kristina.djinovic@univie.ac.at Stephen Graham Division of Structural Biology Wellcome Trust Centre for Human Genetics Roosevelt Drive Oxford OX3 7BN, United Kingdom stepheng@strubi.ox.ac.uk David Stuart Division of Structural Biology Wellcome Trust Centre for Human Genetics Roosevelt Drive Oxford OX3 7BN, United Kingdom dave@strubi.ox.ac.uk Paul Tucker EMBL Hamburg Outstation c/o DESY, Notkestrasse 85 ,D22603 Hamburg, Germany tucker@embl-hamburg.de Nicola Wiskocil Max F. Perutz Laboratories University of Vienna & Medical University Vienna Dr.-Bohrgasse 9/3 A-1030 Wien nicola.wiskocil@univie.ac.at Ursula Thalhammer Department for Biomolecular Structural Chemistry Max F. Perutz Laboratories University of Vienna, Campus Vienna Biocenter 5 A-1030 Vienna, Austria ursula.thalhammer@univie.ac.at Werner Koenig Department for Biomolecular Structural Chemistry Max F. Perutz Laboratories University of Vienna, Campus Vienna Biocenter 5 A-1030 Vienna, Austria werner.koenig@univie.ac.at 2|Page VIZIER / SPINE2 Workshop on Structural Virology PROGRAMME Monday July 14th 2008 11:30 – 13:30 13.30 – 13:40 13:40 - 17:55 Registration, snacks Welcome Session: Entry and Host Interactions Chairs: Dave Stuart, Felix Rey 13:40 – 14:15 Felix Rey Crystal structure of the Rubella virus membrane fusion glycoprotein E1 in its post fusion conformation 14:15 – 14:50 Winfried Weissenhorn Assembly of ESCRT-III and its role in enveloped virus budding 14:50 – 15:20 Eric Huizinga Structures of corona- and torovirus hemagglutininesterases offer insight in receptor specificity and evolution 15:20 – 16:00 Coffee Break 16:00 – 16:35 Katsumi Maenaka Crystal structure of measles virus hemagglutinin provides the molecular basis for effective vaccination 16:35– 17:10 Dieter Blaas Structural Basis of Rhinovirus-Receptor Interaction and Early Processes in Infection 17:10– 17:40 Jan Kadlec Crystal structure of the postfusion form of the baculovirus GP64 protein 17:40 – 18:00 Ulrike Maurer Native 3D analysis of membrane fusion in Herpes simplex virus 1 entry: from a complex virus-host system to single players Free evening 3|Page VIZIER / SPINE2 Workshop on Structural Virology Tuesday July 15th 2008 9:00 - 12:00 Session: Entry and Host Interactions (cont) Chairs: Winfried Weissenhorn 9:00 – 9:35 Jon Grimes TBA 9:35– 10:00 Mark Van-Raaij Structure of avian reovirus fibre Sigma-C and of its double-stranded RNA-binding protein Sigma-A 10:00– 10:25 Eike Schultz Host Recognition of Bacteriohphage K1F: The product complex of EndoNF 10:25 – 11:00 Coffee Break 11:00 - 12:20 Session: Replication Chairs: Bruno Canard 11:00 – 11:35 Stephen Cusack Structure-function relationships of three domains of influenza virus polymerase subunit PB2 11:35 – 12:10 Hongmin Li Structure and function of flavivirus NS5 methyltransferase 12:10 – 12:45 Martino Bolognesi Recognition of RNA cap analogues by Wesselsbron virus NS5 methyl-transferase domain. Implications for flaviviral RNA capping mechanism 12:45 - 15:00 Lunch break and Poster session 15:00 – 15:55 Session: Replication (cont) Chairs: Paul Tucker 15:00 – 15:35 Rolf Hilgenfeld Proteases of RNA viruses and retroviruses: Structures, inhibitors, and resistance 15:35 – 16:10 Bruno Canard The Structural Enzymologist's Gold Mine: The RNA Replication Machinery of Nidovirales 16:10 – 16:30 Mario Milani Structural based inhibition of flavivirus replication enzymes: Helicase and Methyltransferase 16:30 – 17:00 Coffee Break 4|Page VIZIER / SPINE2 Workshop on Structural Virology 17:00 – 18:10 Session: Assembly and Future Perspectives Chairs: Martino Bolognesi 17:00 – 17:35 Rob Ruigrok Structure of negative strand RNA virus nucleocapsids 17:35 – 18:10 Stephen Graham The structures of two rhabovirus matrix (m) proteins reveal a novel mode of self association 20:00 Workshop dinner: Wiener Rathauskeller 5|Page VIZIER / SPINE2 Workshop on Structural Virology Wednesday July 16th 2008 9:30: 12:50 Session: Assembly and Future Perspectives Chairs: Stephen Graham, Rolf Hilgenfeld 09:30 – 10:05 Dennis Bamford Virion architecture and packaging system as probes for virus evolution and classification 10:05 – 11:40 Alasdair Steven DNA Packaging and Capsid-based Signaling as Regulators of Virus Maturation 10:40 – 11:20 Coffee break 11:20 – 11:55 Herman van Tillbeurgh VIRAR : a structural genomics project on archaeal virus proteins 11:55 – 12:30 Gerard Bricogne Some recent developments in instrumentation, protocols and software aimed at challenging structure determination projects 12:30 – 12:40 Concluding remarks 12:40 – 14:00 Lunch 6|Page VIZIER / SPINE2 Workshop on Structural Virology ABSTRACTS OF INVITED SPEAKERS 7|Page VIZIER / SPINE2 Workshop on Structural Virology CRYSTAL STRUCTURE OF THE RUBELLA VIRUS MEMBRANE FUSION GLYCOPROTEIN E1 IN ITS POST-FUSION CONFORMATION. Rebecca Phillips, Marie-Christine Vaney, Rana Al-Kurdi, Félix A. REY Unité de Virologie Structurale, CNRS URA 3015 Department de Virologie, Institut Pasteur 25 rue du Dr. Roux, 75015 Paris France We will present the very recently determined crystal structure of the whole ectodomain of the Rubella virus (RV) glycoprotein E1 at 1.8Å resolution. We produced the recombinant protein in Drosophila Schneider 2 (S2) cells, and we observed that a substantial fraction spontaneously adopts a trimeric, post-fusion conformation. In size exclusion chromatography experiments, about 80% of E1 elutes as trimer, and 20% as monomer. The purified trimers bind to liposomes of various compositions, resulting in a closed-packed hexagonal lattice of trimers coating the surface of the liposomes, independent of the pH. The 3D structure of the RVE1 trimer shows that, as expected, it is a class II membrane fusion protein homologous to alphavirus E1 and flavivirus E. The latter, however, in spite of belonging to two different virus families, appear to be more similar to each other than they are to RVE1. In particular, RVE1 domain II is more elaborate, and several insertions contribute to the formation of two apposed 4-stranded beta sheets. An insertion in the cd loop (which is the fusion loop in class II proteins) in RVE1 results in two distinct loops - separated by about 40 intervening amino acids - located at one end of the trimer. This end is therefore predicted to interact with the target membrane. These two putative fusion loops are rich in glycine and aromatic residues. One of them corresponds to the previously identified “hydrophobic domain” postulated to be the RVE1 fusion loop (Qiu et al, 2000, J Virol 74:6637-6642). Domains I and III do not have any insertions to the standard class II fold. In the trimer, domain III is in a similar orientation as in the post-fusion form of the alphaand flavivirus counterparts, except that its swaps protomers such that domain III from one subunit interacts tightly with domain II of a neighboring subinit in the trimer. The C-terminal stem region – which connects to the TM region - extends all the way toward the membrane to reach the fusion-loop end, making an additional strand enlarging domain II from the neighboring subunit in the trimer. The structure and the comparison to other class II proteins will be presented in this talk. 8|Page VIZIER / SPINE2 Workshop on Structural Virology ASSEMBLY OF ESCRT-III AND ITS ROLE IN ENVELOPED VIRUS BUDDING Suman Lata1, Guy Schoehn1,2, Ankur Jain3, Ricardo Pires1, Jacob Piehler3, Heinrich G. Gőttlinger4 and Winfried WEISSENHORN1 1Unit for Virus Host Cell Interaction, UMR 5233 UJF-EMBL-CNRS, 6 rue Jules Horowitz 38042 Grenoble cedex 9, France 2Institut de Biologie Structurale Jean-Pierre UMR 5075 CEA-CNRS-UJF, 41, rue Jules Horowitz, 38027 Grenole cedex 01, France 3Institute of Biochemistry, Johann Wolfgang Goethe-University, Frankfurt/Main, Germany 4Program in Gene Function and Expression, Program in Molecular Medicine, University of Massachusetts Medical School, Worcester, MA 01605, USA The ESCRT-III complex, formed by CHMP family member proteins, is recruited to membranes and functions at budding steps during multivesicular body biogenesis, cytokinesis and enveloped virus release. Notably enveloped viruses hijack the ESCRT machinery to obtain access to ESCRT-III and the AAA-type ATPase VPS4 to catalyze their release from host cell membranes. We show that ESCRT-III CHMP3 folds into an elongated helical structure that exposes polymerization and membrane targeting surfaces, which are important for HIV-1 budding. At least CHMP3 exists in a soluble closed conformation and an open conformation that probably represents the membrane active form. Co-incubation of C-terminally truncated CHMP2A and CHMP3 results in the assembly of helical tubular structures in vitro. We applied the iterative helical real space reconstruction algorithm to reconstruct volumes from the helical tubular structures. This revealed a structure with a 32 Å helical pitch, containing 16.57 repeating units per turn and inner and outer diameters of 43 nm and 52 nm, respectively; this low resolution structure fits the model of a CHMP2A/CHMP3 heterodimer. We show further that VPS4 binds on the inside of the tubes via the C-terminal region of the CHMP3 and disassembles the tubular structures upon ATP hydrolysis in vitro. In contrast the membrane binding surface of the polymer is positioned on the outside. Our data suggests a model where an ESCRT-III helical structure assembles on the inside of an evolving bud; VPS4 interaction may then control CHMP composition and the diameter of ESCRT-III ring assemblies. Thus the combined function of ESCRT-III and VPS4 might lead to membrane constriction similarly to the mode of action of dynamin in endocytotic membrane fission steps. 9|Page VIZIER / SPINE2 Workshop on Structural Virology CRYSTAL STRUCTURE OF MEASLES VIRUS HEMAGGLUTININ PROVIDES THE MOLECULAR BASIS FOR EFFECTIVE VACCINATION Takao Hashiguchi, Mizuho Kajikawa, Makoto Takeda, Kimiko Kuroki, Yusuke Yanagi, Katsumi MAENAKA Department of Virology, Faculty of Medicine Medical Institute of Bioregulation, Kyushu University 3-1-1 Maidashi, Higashi-ku, Fukuoka, Fukuoka 812-8582 Japan Worldwide, measles virus (MV) still causes 4% of all deaths in children under 5 years of age despite the availability of efficacious vaccines. Unlike many other paramyxoviruses, MV uses signalling lymphocyte activation molecule (SLAM), rather than sialic acid, as a cellular receptor. CD46 also acts as an additional receptor for vaccine strains of MV. A better understanding of MV’s interaction with receptors will facilitate the design of novel strategies for the prevention and treatment of measles infection. Here we report the crystal structure of the MV receptor-binding attachment glycoprotein hemagglutinin (MV-H), which directly recognizes SLAM. Our study reveals an unexpected sugar-shielded structure for MV-H. This conformation renders large areas of the protein inaccessible to both receptor and neutralizing antibodies. Limited surface areas of MV-H are able to act as neutralizing antibody epitopes, one of which corresponds to the SLAM-binding site crucial for MV entry. N-linked sugars also appear to tilt the relative orientation of the dimeric MV-H head domains such that the receptor-binding sites are readily accessible to the cellular receptors. MV vaccines are highly successful; the progenies of the first MV isolate obtained a half century ago are still in use as live vaccines, and no escape mutant strains have been reported. Our structural study clearly revealed that sugar shields of MV-H appear to account for one serotype property of MV, ensuring an effective immune response, in contrast to the sugars of HIV gp120 which allow for immune evasion. Thus, our results suggest the potential ability of sugar shields of microbial glycoproteins to modulate the immune responses by directing them to restricted regions, which may be exploited as a useful tool for vaccine design. 10 | P a g e VIZIER / SPINE2 Workshop on Structural Virology STRUCTURAL BASIS OF RHINOVIRUS-RECEPTOR INTERACTION AND EARLY PROCESSES IN INFECTION Dieter BLAAS Department of Medical Biochemistry Max F. Perutz Laboratories (MFPL) Medical University of Vienna Dr.-Bohr-Gasse 9/3, 1030 Wien Austria Human rhinoviruses (HRVs) are a main cause of upper respiratory infections that usually take a mild course. However, HRVs can also attain the lungs and become life-threatening when other conditions such as asthma, chronic obstructive pulmonary disease and otitis media are pre-existing. The more than 120 HRV types currently known are divided into subgenera A, B, and (tentatively) C. Independent from this classification and despite high sequence and structure similarity HRVs use intercellular adhesion molecule 1 (ICAM-1), low-density lipoprotein receptors (LDLR, VLDLR, and LRP), heparan sulphate, and other, so far unknown receptors for cell entry. This distinction is only partly understood on the molecular basis but comparisons of HRV 3-D structures and sequences of the capsid proteins yield hints as to what might determine receptor selectivity. Furthermore, the receptors also control the entry pathway and mode of uncoating (i.e. release of the genomic RNA into the cytosol). I shall give a summary on the current knowledge on the early stages of HRV infection i.e. virus-receptor interaction, cell surface attachment, entry, and RNA transport through cellular membranes. 11 | P a g e VIZIER / SPINE2 Workshop on Structural Virology CRYSTAL STRUCTURE OF THE POSTFUSION FORM OF THE BACULOVIRUS GP64 PROTEIN Jan KADLEC WTCHG University of Oxford Roosevelt Drive OX3 7BN Oxford United Kingdom 12 | P a g e VIZIER / SPINE2 Workshop on Structural Virology To Be Announced Jonathan GRIMES WTCHG University of Oxford Roosevelt Drive OX3 7BN Oxford United Kingdom 13 | P a g e VIZIER / SPINE2 Workshop on Structural Virology STRUCTURE-FUNCTION RELATIONSHIPS OF THREE DOMAINS OF I NFLUENZA VIRUS POLYMERASE SUBUNIT PB2 Stephen CUSACK EMBL Grenoble Outstation UJF-EMBL-CNRS Unit of Virus Host Cell Interactions 6 rue Jules Horowitz, 38042 Grenoble cedex 9 France Using a novel high-throughput screening technique, called ESPRIT, to experimentally find soluble domains in multi-domain proteins we have identified and determined the crystal structures of three independently folded domains from the polymerase PB2 subunit of influenza A virus. These include a small domain involved in nuclear import of PB2 (1), a cap-binding domain involved in the cap-snatching mechanism of viral transcription (2) and a third domain containing important hostspecific determinants. The structures and their functional implications will be presented. (1) (2) Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Tarendeau F, Boudet J, Guilligay D, Mas PJ, Bougault CM, Boulo S, Baudin F, Ruigrok RW, Daigle N, Ellenberg J, Cusack S, Simorre JP, Hart DJ. Nat Struct Mol Biol. 2007, 14(3):229-33. The structural basis for mRNA cap-binding by influenza virus polymerase subunit PB2. Delphine Guilligay, Franck Tarendeau, Patricia Resa-Infante, Rocío Coloma, Thibaut Crepin, Rob W. H. Ruigrok, Juan Ortin, Darren J. Hart and Stephen Cusack. Nat. Struct. Mol. Biol. 2008, 15(5):500-506. The work has been partly financed by the EU FLUPOL contract (SP5B-CT-2007-044263). 14 | P a g e VIZIER / SPINE2 Workshop on Structural Virology STRUCTURE AND FUNCTION OF FLAVIVIRUS NS5 METHYLTRANSFERASE Hongmin LI Wadsworth Center New York State Department of Health 120 New Scotland Ave, Albany, NY 12208 USA The plus-strand RNA genome of flavivirus contains a 5' terminal cap 1 structure 7 (m GpppAmG). The flaviviruses encode one methyltransferase (MTase), located at the N-terminal portion of the NS5 RNA-dependent RNA polymerase (RdRp). The NS5 MTase catalyzes both guanine N-7 and ribose 2'-OH methylations during viral cap formation (1, 2). Representative flavivirus MTases from dengue, yellow fever, 7 7 and West Nile virus (WNV) sequentially generate GpppA-->m GpppA-->m GpppAm (3, 4). Despite exhibiting two distinct methylation activities, the crystal structures of flavivirus MTases showed a single binding site for S-adenosyl-L-methionine (SAM), the methyl donor (1, 3, 5). This indicates that substrate GpppA-RNA should be repositioned to accept the N-7 and 2'-O methyl groups from SAM during the sequential reactions. Further studies demonstrated that distinct RNA elements are required for the methylations at guanine N-7 on the cap and ribose 2'-OH on the first transcribed nucleotide (4). In a WNV model, N-7 cap methylation requires specific nucleotides at the second and third positions and a 5' stem-loop structure; in contrast, 2'-OH ribose methylation requires specific nucleotides at the first and second positions, with a minimum 5' viral RNA of 20 nucleotides. Mutant enzymes with different methylation defects can trans complement one another in vitro, demonstrating that separate molecules of the enzyme can independently catalyze the two cap methylations in vitro (6, 7). Mutation analysis further demonstrated that defects in the MTase domain of NS5 can be compensated by mutations in the RdRp domain, indicating a direct correlation between the MTase and RdRp functions of the NS5 protein to complete virus replication (8-10). In the context of complete virus, defects in both methylations are lethal to WNV; however, viruses defective solely in 2'-O methylation are attenuated and can protect mice from later wild-type WNV challenge (2, 3). The results demonstrate that the N-7 methylation activity is essential for the WNV life cycle and, thus, methyltransferase represents a novel target for flavivirus therapy. 1) Egloff, M. P., D. Benarroch, B. Selisko, J. L. Romette, and B. Canard. 2002. An RNA cap (nucleoside-2'-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. Embo J 21:2757. 2) Ray, D., A. Shah, M. Tilgner, Y. Guo, Y. Zhao, H. Dong, T. Deas, Y. Zhou, H. Li, and P. Shi. 2006. West nile virus 5'-cap structure is formed by sequential guanine N-7 and ribose 2'-O methylations by nonstructural protein 5. J. Virol. 80:8362. 3) Zhou, Y., D. Ray, Y. Zhao, H. Dong, S. Ren, Z. Li, Y. Guo, K. A. Bernard, P. Y. Shi, and H. Li. 2007. Structure and function of flavivirus NS5 methyltransferase. J Virol 81:3891. 4) Dong, H., D. Ray, S. Ren, B. Zhang, F. Puig-Basagoiti, Y. Takagi, C. K. Ho, H. Li, and P. Y. Shi. 2007. Distinct RNA elements confer specificity to flavivirus RNA cap methylation events. J Virol 81:4412. 5) Egloff, M. P., E. Decroly, H. Malet, B. Selisko, D. Benarroch, F. Ferron, and B. Canard. 2007. Structural and Functional Analysis of Methylation and 5'-RNA Sequence Requirements of Short Capped RNAs by the Methyltransferase Domain of Dengue Virus NS5. J Mol Biol 372:723. 15 | P a g e VIZIER / SPINE2 Workshop on Structural Virology 6) Dong, H., S. Ren, H. Li, and P. Y. Shi. 2008. Separate molecules of West Nile virus methyltransferase can independently catalyze the N7 and 2'-O methylations of viral RNA cap. Virology 377:1. 7) Dong, H., S. Ren, B. Zhang, Y. Zhou, F. Puig-Basagoiti, H. Li, and P. Y. Shi. 2008. West Nile virus methyltransferase catalyzes two methylations of the viral RNA cap through a substraterepositioning mechanism. J Virol 82:4295. 8) Zhang, B., H. Dong, Y. Zhou, and P. Y. Shi. 2008. Genetic interactions among the West Nile virus methyltransferase, the RNA-dependent RNA polymerase, and the 5' stem-loop of genomic RNA. J Virol. 9) Yap, T. L., T. Xu, Y. L. Chen, H. Malet, M. P. Egloff, B. Canard, S. G. Vasudevan, and J. Lescar. 2007. Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J Virol 81:4753. 10) Malet, H., M. P. Egloff, B. Selisko, R. E. Butcher, P. J. Wright, M. Roberts, A. Gruez, G. Sulzenbacher, C. Vonrhein, G. Bricogne, J. M. Mackenzie, A. A. Khromykh, A. D. Davidson, and B. Canard. 2007. Crystal structure of the RNA polymerase domain of the West Nile virus non-structural protein 5. J Biol Chem 282:10678. 16 | P a g e VIZIER / SPINE2 Workshop on Structural Virology RECOGNITION OF RNA CAP ANALOGUES BY WESSELSBRON VIRUS NS5 METHYLTRANSFERASE DOMAIN. IMPLICATIONS FOR FLAVIVIRAL RNA CAPPING MECHANISM Eloise Mastrangelo, Mario Milani, Michela Bollati, Graziella Sorrentino and Martino BOLOGNESI Department of Biomolecular Sciences and Biotechnology University of Milan Via Celoria 26, 20133 Milan Italy Viral methyltransferases are involved in completion of the mRNA capping process, transferring a methyl group from S-adenosyl-L-methionine to capped RNA. The Flavivirus genome is capped at the 5’ terminus with the “cap I” structure, that is essential for mRNA stability and proper replication. N7- and 2’O-methyltransferase activities have been recently associated to the N-terminal domain of the viral NS5 protein. In order to further characterize the series of enzymatic reaction supporting capping, our lab analyzed several crystal structures of Wesselsbron virus MTase, in complexes with the AdoMet cofactor, with AdoHcy (the product of the methylation reaction), with Sinefungin (a molecular analogue of AdoMet), and with three different cap-analogues (GpppG, N7MeGpppG, N7MeGpppA; Bollati et al. in preparation). The different crystal structures show that the 5’ Guanine is constantly bound to a “high affinity” site region located next to the N-terminal region of the enzyme. Conformational disorder affects systematically the 3’ end of the cap analogues, following the second or the third phosphate of the ppp bridge. The N7-methyl group does not display any contact with the protein, being solvent accessible; on the other hand, electrostatic effects related to N7-methylation may play a recognition role. Following incubation with GTP, Wesselsbron MTase displays a GMP molecule covalently bound to residue Lys28, although to a fraction of the whole molecular population; such an observation may suggest implications for transfer of a guanine group to the ppRNA. The structures of the MTase complexes obtained and the binding assays are discussed in the context of a model for N7-MTase and 2’OMTase activities. This work has been developed in close collaboration with Laboratoire Architecture et Fonction des Macromolécules Biologiques (Marseille, France) 17 | P a g e VIZIER / SPINE2 Workshop on Structural Virology PROTEASES OF RNA VIRUSES AND RETROVIRUSES: STRUCTURES, INHIBITORS, AND RESISTANCE Rolf HILGENFELD Institute of Biochemistry Center for Structural and Cell Biology in Medicine (CSCM) University of Lübeck Ratzeburger Allee 160, 23538 Lübeck Germany Viral proteases are important targets for structure-based drug discovery. Prime examples are the proteinases of coronaviruses and picorna- as well as enteroviruses, and the retroviral proteases, Progress over the past few years in developing inhibitors for these targets will be described. The development of viral resistance against drugs seems to be inevitable. The structural evolution of resistance mutants of the HIV-1 proteinase in a patient treated with ritonavir will be discussed and efforts to overcome such resistance will be presented. It will be suggested to abandon the dogma that the best inhibitors are those offering maximum interaction between their side chains and the target enzyme. References: 1) J. Weber, J.R. Mesters, M. Lepsik, J. Prejdova, M. Svec, J. Sponarova, P. Mlcochova, K. Skalicka, K. Strisovsky, T. Uhlikova, M. Soucek, L. Machala, M. Stankova, J. Vondrasek, T. Klimkait, H.-G. Kraeusslich, R. Hilgenfeld & J. Konvalinka: Unusual binding mode of an HIV-1 protease inhibitor explains its potency against multi-drug-resistant virus strains. J. Mol. Biol. 324, 739-754 (2002). 2) K. Anand, G.J. Palm, J.R. Mesters, S.G. Siddell, J. Ziebuhr & R. Hilgenfeld: Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 21, 3213-3224 (2002). 3) K. Anand, J. Ziebuhr, P. Wadhwani, J.R. Mesters & R. Hilgenfeld: Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 300, 1763-1767 (2003). 4) H. Yang, M. Yang, Y. Ding, Y. Liu, Z. Lou, Z. Zhou, L. Sun, L. Mo, S. Ye, H. Pang, G.F. Gao, K. Anand, M. Bartlam, R. Hilgenfeld & Z. Rao: The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 100, 13190-13195 (2003). 5) J. Tan, K.H.G. Verschueren, K. Anand, J. Shen, M. Yang, Y. Xu, Z. Rao, J. Bigalke, B. Heisen, J. Mesters, K. Chen, X. Shen, H. Jiang & R. Hilgenfeld: pH-dependent conformational flexibility of the SARS-CoV main proteinase (Mpro) dimer: Molecular dynamics simulations and multiple X-ray structure analyses. J. Mol. Biol. 354, 25-40 (2005). 6) J.R. Mesters, J. Tan & R. Hilgenfeld: Viral enzymes. Curr. Opin. Struct. Biol. 16, 776-786 (2006). 7) K.H.G. Verschueren, K. Pumpor, S. Anemüller, S. Chen, J.R. Mesters & R. Hilgenfeld: A structural view of the inactivation of the SARS coronavirus main proteinase by benzotriazole esters. Chem. Biol. 15, 597-606 (2008). 18 | P a g e VIZIER / SPINE2 Workshop on Structural Virology THE STRUCTURAL ENZYMOLOGIST'S GOLD MINE: THE RNA REPLICATION MACHINERY OF NIDOVIRALES Bruno CANARD Architecture and Function of Biological Molecules CNRS-Université AIX-Marseille I & II 163, Av de Luminy, 13288 Marseille France The order Nidovirales is comprised of the Coronaviridae (Coronavirus and Torovirus), the Arteriviridae, and the Roniviridae. They infect animals and man, and the resulting pathology can range from benign to fatal. A common feature of the order is a complex organisation of both gene products and gene expression. Nidovirales have in common a polycistronic genome, a large replicase open reading frame whose proteolytic product stoechiometry is controlled by a frameshift before the RNA dependent RNA polymerase gene cluster, and the ability to synthesize a set of nested subgenomic RNAs encoding sructural proteins. In 2003, the emergence of a Severe Acute Respiratory Syndrome due to a Coronavirus (SARS-CoV) has dramatically shown the scarcity of biochemical and structural data available regarding Nidovirales. The Nidovirales replication machinery is however an unbelievable treasure for the enzymologist. The main replicative ORF encodes up to 16 multifunctional proteins involved in sophisticated coordinated RNA replication/transcription. Interestingly, many putative enzyme activities are coronavirus-unique and not directly related to "traditional" viral RNA replication. Although some proteins remain with no putative function associated, many enzyme activities have been predicted using bio-informatic approaches, and the corresponding activities demonstrated. There are however a large number of question marks regarding the role these activities play in any of the processes occuring during a Nidovirales life-cycle. There are a number of possibilities regarding the involvement of the different orf1a and b partners. The presence of seemingly RNA replication-irrelevant enzyme activities, such as the macro domain of Coronavirus nsp3, argues in favor of a role into counteracting the host defence mechanisms, whereas the presence of endo and exonuclease activities suggests the presence of RNA repair systems or microRNA production machineries absent in other (+)RNA genome viruses. We have engaged into a structural and functional study of the Nidovirales order in order to cast light into this most interesting and sophisticated RNA replication machinery. We have reported, for coronavirus, the crystal structure at 2.8 Å resolution of Nsp9, an RNA SSB protein unique in the RNA virus world, that may have a role in several processes such as the regulation of SARS-CoV replication/transcription. Likewise, the structure of the histone macro domain protein has been solved at 1.8 Å resolution, and shown to carry an ADP-1"-phosphate hydrolase activity, usually occuring in tRNA splicing pathways. The NendoU endonuclease hexameric structure has been determined at 2.6 Å resolution and shown to carry an uncommon Mn 2+dependent RNase-A like activity. More recently, we have found that the nsp7/8 complex is endowed with RNA-dependent RNA primase activity, a feature consistent with the classification of primer-dependent replication of Nidovirales in the 19 | P a g e VIZIER / SPINE2 Workshop on Structural Virology Picornavirus superfamily. The last gene product of the orf1b has been demonstrated to be a Cap-0 dependent 2'-O-methyltransferase, resembling that of DNA viruses rather than the phylogenetically closer Flavivirus. In Arteriviridae, we have demonstrated that the nsp9 protein is an RNA dependent RNA polymerase, and that a primase system resembling that of the coronavirus might be present as well. I will present the current status of the coronavirus replicase complex based on current structural and enzymatic results. Although the SARS crisis has acted as a powerful boost in structural and functional studies, it is essential to re-affirm that the whole Nidovirales order is of equal scientific interest, and that fundamental knowledge acquired on this order is also expected to bring stunning discoveries translastable into efficient drug design should a new Nidovirus emerge in animal or man. 20 | P a g e VIZIER / SPINE2 Workshop on Structural Virology Structure of negative strand RNA virus nucleocapsids Rob RUIGROK, Marc Jamin and Martin Blackledge Unit for Virus Host Cell Interactions UJF-EMBL-CNRS UMR 5233 Institut de Biologie Structurale Jean-Pierre Ebel CEA; CNRS; UJF UMR 5075 Grenoble France Non-segmented negative strand RNA viruses have their viral RNA tightly associated with the viral nucleoprotein. Such N-RNA complexes form irregular and flexible helices. The structure of recombinant N-RNA from rabies virus and vesicular stomatitis virus (VSV) was determined at atomic resolution. Nucleocapsids from other viral families have only been studied by EM. The viral polymerase is attached to these structures through the polymerase cofactor, the phosphoprotein (P). During the last two years we have concentrated our work on the structures of the phosphoproteins from the rhabdovirus (rabies virus and VSV) and paramyxovirus (Sendai virus) families. These proteins all have two or three structured domains (from N- to C-terminus: A domain that binds to RNA-free N (N0), an oligomerisation domain and an N-RNA binding domain) that are connected by disordered or flexible linkers. For Sendai virus we have determined the structure of the N-RNA binding domain of P and we have characterized the flexibility in this domain by NMR. The peptide of N that binds to P (NTAIL) was also identified and by an analysis of NMR Residual Dipolar Couplings we were able to describe the molecular recognition element of NTAIL as a population of three specific overlapping helical conformers. The dynamic structure of both partners may explain the rapidly making and breaking of contacts between P and N in N-RNA during transcription or replication by the polymerase. For rabies virus we have defined the borders of the ordered domains and their function and for VSV we have determined the structure of the N-RNA binding domain. The structure of this domain will be compared with that of rabies virus. The people from our labs and from other labs that have contributed to this work are: Francine Gerard, Euripedes Ribeiro Jr., Malene Ringkjøbing Jensen, Klaartje Houben, Cédric Leyrat, Ivan Ivanov, Ewen Lescop, Laurence Blanchard, Danielle Blondel, Sonia Longhi, Aurélie Albertini, Manos Mavrakis, Bernhard Brutscher, Adrien Favier. 21 | P a g e VIZIER / SPINE2 Workshop on Structural Virology THE STRUCTURES OF TWO RHABDOVIRUS MATRIX (M) PROTEINS REVEAL A NOVEL MODE OF SELF ASSOCIATION Stephen C. GRAHAM1, René Assenberg1, Olivier Delmas2, Anil Verma1, Raymond J. Owens1, David I. Stuart1, Jonathan M. Grimes1 and Hervé Bourhy2 1Division of Structural Biology and Oxford Protein Production Facility, Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford OX3 7BN, UK 2UPRE Lyssavirus Dynamics and Host Adaptation, WHO Collaborating Centre for Reference and Research on Rabies, Institut Pasteur, 75724 Paris CEDEX 15, France The Rhabdoviridiae family of viruses, typified by Rabies virus (a lyssavirus and the etiologic agent of lethal meningoencephalitis) and vesiculostomatitis virus (VSV) possess a negative-sense single-stranded RNA genome that encodes five proteins. The rhabdovirus matrix (M) protein is a multifunctional protein implicated in regulating the switch between transcription and replication of the viral genome, modulating transcription of host genes, and in virus assembly and budding. To date, the only structural information available on rhabdovirus M proteins is the structure of a thermolysin-stable M core of VSV, which lacks the N-terminal 47 residues (including a PPXY ‘late domain’ thought to be essential for virus budding) and a surface-exposed hydrophobic loop due to proteolysis. To clarify the various roles of M throughout the rhabdovirus lifecycle we attempted to solve the crystal structures of full-length lyssavirus and vesiculovirus M proteins. We successfully cloned, expressed and purified the M proteins from three lyssaviruses (Lagos bat virus (LAG), Mokola virus and Thailand dog virus) and two VSV viruses (VSV Indiana and VSV New Jersey). Crystals were obtained for fulllength M proteins of LAG and VSV New Jersey. Successful crystallization depended on a number of factors, in particular the addition of an N-terminal SUMO fusion tag to the protein to increase solubility. Residues 48-202 of LAG M form a globular domain closely resembling VSV M despite the aligned residues sharing only 9% sequence identity. Whilst no electron density is evident for residues 1–30 and 38–47, residues 30–37 (which overlap with the PPEY ‘late domain’ of LAG M; residues 35–38) form a short stretch of lefthanded polyproline-II helix and are bound to a hydrophobic groove on the surface of the globular domain. Strikingly, the structure of VSV M also reveals an ordered segment of the otherwise disordered N-terminus bound to the main globular domain. While the general location of the binding site for the ordered segment in VSV M is similar to that observed for LAG M, the nature of the interaction, including the orientation of the bound segment and the nature of the amino acids at the interface, differs significantly. The implications of these novel self-associations for virus morphogenesis and for mediating interactions between M and host proteins will be discussed. 22 | P a g e VIZIER / SPINE2 Workshop on Structural Virology VIRION ARCHITECTURE AND PACKAGING SYSTEM AS PROBES FOR VIRUS EVOLUTION AND CLASSIFICATION Dennis H. BAMFORD Institute of Biotechnology and Department of Biological and Environmental Sciences University of Helsinki Biocenter 2, P.O. Box 56 Finland Viruses are the most abundant living entities in the biosphere outnumbering their host organisms by one to two orders of magnitude. It is conceivable that they cause the highest selective pressure their hosts encounter. As obligate parasites viruses are dependent on their hosts but their origins seem to deviate from that of cellular life. What are the possible principles to build viruses is an open question. However, structural studies on virus capsids and coat proteins propose that there are only a limited number of ways to construct a viral protein coat. Consequently, relatedness of viruses is not connected to the type of cells they infect and the same architectural principle of the capsid has been observed in viruses infecting bacteria as well as humans. Using the viral capsid structure it is possible to group viruses to several lineages that may have existed before the three cellular domains of life (bacteria, archaea and eukarya) were separated. In addition to the capsid proteins putative (packaging) ATPases form the “self elements” that allow to trace such elements in bacterial, archaeal and eukaryotic genomes. This would mean that viruses are ancient and that early cells were infected with many viruses proposing that the origin of viruses is polyphyletic opposing the monophyletic origin of cellular life. 23 | P a g e VIZIER / SPINE2 Workshop on Structural Virology DNA PACKAGING AND CAPSID-BASED SIGNALING AS REGULATORS OF VIRUS MATURATION. Alasdair C. STEVEN Laboratory of Structural Biology National Institute of Arthritis, Musculoskeletal and Skin Diseases National Institutes of Health Bethesda MD 20892 USA Despite their entirely disparate host ranges, the tailed bacteriophages and the herpesviruses share many features in their capsid assembly pathways, probably reflecting common albeit distant evolutionary origins (1). In both cases, the capsid is first assembled as a precursor procapsid into which the linear double-stranded DNA genome is packaged from a replicating concatemer by a viral ATPase/DNA translocase/endonuclease (aka terminase). This sequence of events must be regulated at several levels: how is packaging initiated? How is it determined that packaging must cease? What signals that a phage nucleocapsid is ready to bind its tail or that a herpesvirus nucleocapsid should exit the nucleus and proceed on its envelopment pathway? This talk will discuss observations on herpes simplex virus type 1 and coliphage HK97 that indicate that, in both cases, a key regulatory event is a relatively subtle conformational change in the capsid shells induced by the pressure of packaged DNA on the inner capsid surface late in the packaging process. This transition is distinct from the relatively dramatic conformational changes that are the hallmark of procapsid maturation but is sufficient to facilitate protein-protein interactions that allow maturation to be completed. 1. Steven, A.C., Heymann, J.B., Cheng, N., Trus, B.L., and Conway, J.F. (2005). Curr. Op. Struct. Biol. 15, 227-236. 24 | P a g e VIZIER / SPINE2 Workshop on Structural Virology VIRAR : A STRUCTURAL GENOMICS PROJECT ON ARCHAEAL VIRUS PROTEINS Consortium group leaders: H. VAN TILBEURGH1, C Cambillau2 and D. Prangishvilli3 1 IBBMC, 2 AFMB, Université de Paris Sud, Orsay, France Université de Aix Marseille 2, CNRS, Marseille, France 3 Institut Pasteur, Paris, France Samples collected from natural habitats that contain predominantly archaea have revealed viruses with totally novel morphotypes. Enormous progress has recently been made in the characterization and genomic sequencing of these ds DNA viruses. Most of the archaeal viruses are unrelated to other known viruses or phages, suggesting that they may have different evolutionary origins. In order to better understand the biology and evolution of these fascinating viruses, we decided to launch a structural proteomics project on a few archaeal virus families. We focused our efforts on viruses infecting Crenarchaeota. We cloned and overexpressed putative proteins from linear particle viruses belonging to the rod-like Rudiviridae and filamentous Lipothrixviridae families. We have obtained up till now 8 new protein structures, most of them with new folds. These structures and some corresponding functional data will be presented. 25 | P a g e VIZIER / SPINE2 Workshop on Structural Virology SOME RECENT DEVELOPMENTS IN INSTRUMENTATION, PROTOCOLS AND SOFTWARE AIMED AT CHALLENGING STRUCTURE DETERMINATION PROJECTS Gérard BRICOGNE Global Phasing Ltd. Sheraton House, Castle Park Cambridge CB3 0AX United Kingdom There are numerous typical difficulties associated with challenging structure determination projects: small crystals, often with multiple lattices and unfavourable morphologies, showing anisotropic diffraction, too radiation sensitive for a single sample to yield a complete dataset, while non-isomorphism between different crystals precludes a simple-minded merging of partial datasets from several crystals. Several ongoing developments have the potential of alleviating these difficulties, a few of which will be described in this talk. A proper statistical analysis of radiation damage effects, capable of representing the gradual drift of diffraction data with increasing radiation dose, is still lacking. When such effects are dominated by site-specific damage, their use for phasing through the available methods is predicated on the assumption that all sources of nonisomorphism are uncorrelated between different observations of a given reflection. A treatment of such damage by a dose-dependent parametrisation of anomalous scatterers [1] overcomes the limitations of this assumption as far as the site-specific component of non-isomorphism is concerned. However, the problem remains for the general damage to the macromolecule, which requires that the foundations of scaling (with or without merging), phasing (in the structure of the likelihood functions) and refinement (of which structure) be re-examined from first principles. This is possible by means of multivariate likelihood functions for arbitrary covariances of uncertainties on complex-valued structure factors [2]. The relevance of this approach to the successive steps of data reduction and structure determination will be discussed. An intuitive and already well-known practical conclusion is that data for SAD or MAD phasing should be collected in such a way that all measurements relevant to the phasing of a given unique reflection be recorded as close in dose as possible. Such experimental designs were used in the early days of the MAD method but have unfortunately been largely overlooked since. Combined with new implementations of kappa goniometry and suitably adapted data processing, they constitute an effective counter-measure against radiation damage. Another option is to increase the amount of phase information extracted per unit dose inflicted on a crystal. It will be shown that the rather obscure phenomenon of AAS (anisotropy of anomalous scattering), once thought to possibly undermine the very viability of the MAD method [3], provides such an opportunity [4]. References: [1] Schiltz, M., Dumas, P., Ennifar, E., Flensburg, C., Paciorek, W., Vonrhein, C. & Bricogne, G., Acta Cryst. D60, 1024-1031, 2004. [2] Bricogne, G., Proceedings of the Workshop on Advanced Special Functions and Applications, Melfi (PZ), Italy, 9-12 May 1999, edited by D. Cocolicchio, G. Dattoli & H. M. Srivastava, pp 315-321. Rome: Aracne Editrice 2000. [3] Fanchon, E. & Hendrickson, W.A., Acta Cryst. A46, 809-820, 1990. [4] Schiltz, M. & Bricogne, G., Acta Cryst. D64, 711-729. 26 | P a g e VIZIER / SPINE2 Workshop on Structural Virology POSTER ABSTRACTS 27 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.01 STRUCTURES OF CORONA- AND TOROVIRUS HEMAGGLUTININ-ESTERASES OFFER INSIGHT IN RECEPTOR SPECIFICITY AND EVOLUTION Martijn A. Langereis2, Qinghong Zeng1, Arno L.W. van Vliet2, Raoul J. de Groot2 and Eric G. HUIZINGA1 1Crystal and Structural Chemistry, Bijvoet Center for Biomolecular Research, Faculty of Science and 2Virology Division, Department of Infectious Diseases & Immunology, Faculty of Veterinary Medicine Utrecht University 1Padualaan 8, 3584 CH Utrecht The Netherlands The hemagglutinin-esterases (HEs) are a family of viral envelope glycoproteins that mediate reversible attachment to O-acetylated sialic acids by acting both as lectins and as receptor-destroying enzymes (RDEs). This dual activity prevents virions from being trapped by receptors located on nonpermissive cells, the heavy sialylated mucins of the mucus barrier, or other virus particles. Related HEs occur in influenza C, toro- and coronaviruses apparently as a result of relatively recent lateral gene transfer events. We determined crystal structures of toro- and coronavirus HEs that display a preference for 9-O- or 7,9-O-acetylated sialic acid. Key to obtaining sufficient amounts of protein suitable for crystallization was the use of codon-optimized plasmids and expression in N-acetylglucosaminyltrans-ferase-I deficient HEK293S cells. The crystal structures show that toro- and coronavirus HEs arose from an influenza C-like hemagglutinin-esterase fusion protein (HEF). In the process, HE was transformed from a trimer into a dimer, while remnants of the fusion domain were adapted to establish new monomer-monomer contacts. The structural design of the RDE-acetylesterase domain remained essentially unaltered. Its catalytic pocket shows limited variation that correlates with differences in substrate preference. In contrast the HE receptor-binding domain underwent extensive remodeling and is markedly different even in HEs with shared specificity: the 9-O specific corona- and torovirus HEs bind the receptor in an orientation that is reversed with respect to 9-O specific HEF. The enormous variation in the receptor binding site is surprising as the architecture of the HEF site was preserved in influenza A hemagglutinin (HA) over a much larger evolutionary distance. Apparently, HA and HEF are under more stringent selective constraints than HE, limiting their exploration of alternative binding-site topologies. We attribute the plasticity of the CoV HE receptorbinding site to evolutionary flexibility conferred by functional redundancy between HE and its companion spike protein S. Our findings offer unique insights into the structural and functional consequences of independent protein evolution following interviral gene exchange and open new potential avenues to broad-spectrum antiviral drug design. 28 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.02 HOST RECOGNITION OF BACTERIOHPHAGE K1F: THE PRODUCT COMPLEX OF EndoNF Eike - Christian SCHULZ1, Katharina Stummeyer2, Achim Dickmanns1, Rita Gerardy-Schahn2 and Ralf Ficner1 1: Abteilung für Molekulare Strukturbiologie, Institut für Mikrobiologie und Genetik, Georg-August-Universität Göttingen, Justus-von-Liebig-Weg 11, 37077 Göttingen, Germany 2: Medizinische Hochschule Hannover, MHH, Abteilung Zellulaere Chemie, OE 4330 Carl-Neuberg-Strasse 1, 30625 Hannover, Germany Alpha-2,8-linked polysialic acid (polySia) is an important mediator of cellular motility and functional plasticity in the vertebrate brain and has implications in tumor metastasis. PolySia is also a common cell wall modification of pathogenic prokaryotes like Escherichia coli K1 and Neisseria meningitides serogroup B that cause meningitis and severe sepsis in humans. The cell wall modification serves as a protective barrier against harmful influences such as viral infections. The only known source for enzymes that specifically degrade polySia are E. coli K1 specific bacteriophages. They carry endosialidases as host specificity determining tailspike proteins required to digest the bacterial polySia capsule during infection. We now determined three crystal structures of active site mutants of the endosialidase cloned from bacteriophage K1F (endoNF) in complex with oligomeric sialic acid. The structures have been refined to resolutions up to 1.4 Å. A well defined electron density map of oligomeric sialic acid could be observed for three binding sites, one of which is located in the active site cleft. The complex structures confirm the helical conformation of α-2,8-linked polySia and allow new mechanistic conclusions. 29 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.03 NATIVE 3D ANALYSIS OF MEMBRANE FUSION IN HERPES SIMPLEX VIRUS 1 ENTRY: FROM A COMPLEX VIRUS-HOST SYSTEM TO SINGLE PLAYERS Ulrike E. MAURER°, Florent C. Bender*, J. Charles Whitbeck*, Brian Hannah*, Roselyn J. Eisenberg*, Gary H. Cohen*, Juha T. Huiskonen §°, Beate Sodeik# , Kay Grünewald° *Department of Microbiology, School of Dental Medicine, and Department of Pathobiology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA 19104. °Department of Molecular Structural Biology, Max-Planck-Institute of Biochemistry, Martinsried, Germany §Institute of Biotechnology, University of Helsinki, Finland #Institute of Virology, Hannover Medical School, Germany Glycoprotein B (gB) from herpes simplex virus 1 (HSV-1) is an essential envelope protein that is highly conserved among the members of the Herpesviridae. Together with gD and gH/gL, it mediates the entry of the virus into host cells. Recently, we captured in situ snapshots of the virus fusion at the plasma membrane from adherent cells and synaptosomes using cryo electron tomography [1]. The incoming capsid separated from the tegument and was closely surrounded by the cortical cytoskeleton. After entry, the viral membrane curvature changed concomitantly with a reorganization of the envelope glycoprotein spikes. Individual glycoprotein intermediates were revealed in different conformational stages during entry, thus showing the complex viral fusion mechanism in action. To further analyze the structural changes occurring in gB domains in the course of interaction with the membrane, we incubated the soluble truncated form of gB (gB730t, residues 31-730) with liposomes. This form of gB lacks the transmembrane domain, but was still able to bind to liposomes, and completely covered the liposome surface. We determined the structure of liposome-bound gB730t by averaging subvolumes extracted from the tomographic reconstructions. By fitting the x-ray structure of gB [2] into our structure, we were able to determine the lipid binding site. We are currently expanding this approach by including other truncated forms of gB as well as gB mutants to further characterize functional domains of the protein, which play a crucial role in membrane fusion during HSV-1 entry. [1] [2] Maurer UE, Sodeik B and Grünewald K (2008) Native 3D Intermediates of Membrane Fusion in Herpes Simplex Virus 1 Entry. PNAS (accepted) Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ and Harrison SC (2006) Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217-220. 30 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.04 STRUCTURES OF AVIAN REOVIRUS FIBRE SIGMA-C AND OF ITS DOUBLE-STRANDED RNA-BINDING PROTEIN SIGMA-A Pablo Guardado-Calvo1, X. Lois Hermo-Parrado1, Gavin C. Fox3, Antonio L. LlamasSaiz2, José Martinez-Costas1, J. Benavente1, Mark J. VAN RAAIJ1,4 1Department of Biochemistry and Molecular Biology and 2X-ray Unit, University of Santiago de Compostela, E-15782 Santiago de Compostela, Spain 3Spanish CRG Beamline BM16, European Synchrotron Radiation Facility, 6 rue Jules Horowitz, F-38043 Grenoble, France 4Department of Structural Biology, Institute of Molecular Biology of Barcelona-CSIC, c/Josep Samitier 1-5, E-08028 Barcelona, Spain Avian reovirus fibre, a homo-trimer of the sigmaC protein, is responsible for primary host cell attachment. Partial proteolysis yielded a C-terminal protease-stable receptor-binding domain that could be crystallised. We have solved its structure using two-wavelength anomalous diffraction. The C-terminal globular domain has a beta-barrel fold with the same overall topology as mammalian reovirus fibre (sigma1). The monomers show a more splayed-out arrangement than in the sigma1 structure. Also resolved are two triple beta-spiral repeats of the stalk. Recently, the structure could be extended further towards the N-terminus, showing the shaft domain also contains a triple alpha-helical coiled coil. The avian reovirus protein sigmaA plays a dual role, it is a structural protein, part of the transcriptionally active core, but it is also implicated in the virus' resistance to interferon by binding dsRNA and thus preventing the activation of the dsRNAdependent protein kinase. We have crystallised the protein in absence of RNA and solved its structure by molecular replacement, using the mammalian reovirus sigma2 structure. Twelve crystallographically independent molecules were located in the P1 unit cell. The supramolecular structure suggests a single-helical model for dsRNA binding; site-directed mutagenesis, analytical ultracentrifugation and electron microscopy results are consistent with this model. 31 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.05 STRUCTURAL BASED INHIBITION OF FLAVIVIRUS REPLICATION ENZYMES: HELICASE AND METHYLTRANSFERASE Mario MILANI1, Eloise Mastrangelo1, Michela Bollati1, Etienne Decroly2, and Martino Bolognesi1 1Department of Biomolecular Sciences and Biotechnology, CNR-INFM, University of Milano, Via Celoria 26, 20133-Milano, Italy Architecture et Fonction des Macromolécules Biologiques, UMR 6098, AFMB-CNRS-ESIL, Case 925, 163 Avenue de Luminy, 13288-Marseille, France Unité des Virus Emergents, Faculté de Médecine, 27 Bd Jean Moulin, 13005Marseille, France 2Laboratoire The genus Flavivirus comprises over seventy RNA viruses, many of which are important human pathogens. The Flavivirus genome consists of 11 kb capped at the 5’ terminus encoding a 370 kDa polyprotein precursor. The polyprotein is processed into three structural proteins and seven non structural proteins involved in the viral replication (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) (Fields et al., 2001). During viral replication the nascent transcripts must be unwound from their complementary template RNA: such role is displayed by the C-terminal domain of the NS3 protein (helicase, Hel domain). In addition, proper translation requires the capping of viral RNA and among other enzymes is essential the function of the NS5 protein N-terminal domain (methyltransferase activity, Mtase). Consequently, the inhibition of flaviviral Hel and Mtase might provide a tool against flaviviral infection. In this work we used virtual docking approach (autodock4) to examine the affinity of various commercial compounds against the two viral enzymes. After identification of the best candidate inhibitors, the compounds have been tested by in vitro activity/inhibition assays showing IC50 from 2 to 0.01 uM. The development of mechanistic hypothesis for the activity of both the enzymes, which we based on our previous crystal structure analysis, was instrumental to the achievement of these results. In fact, based on such hypothesis, novel sites for inhibitor binding could be proposed and subsequently validated through the above described approach 32 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.06 Structural study of human astrovirus protease and Rabbit hemorrhagic disease virus protease Daniele BONIVENTO1, Silvia Speroni1, Jacques Rohayem2, Simone Nenci1, Bruno Coutard3, Bruno Canard3, and Andrea Mattevi1 1 Dept. of Genetics and Microbiology, University of Pavia, via Ferrata 1, Pavia, 27100 Italy 2 The Calicilab, Institute of Virology, Dresden University of Technology, Fiedlerstrasse 42, 01307 Dresden, Germany 3AFMB UMR 6098 CNRS/UI/UII ESIL Case 932 163 Avenue de Luminy 13288 Marseille cedex 9, France Different classes of RNA viruses adopt a translational strategy based on the proteolytic processing of a polyprotein, with subsequent release of the viral replicative enzymes. The proteases involved in the processing of the polyprotein thus constitute a key element in the post-translational regulation of the viral machinery and represent relevant drug targets. In order to gain more insight into the mode of action of these enzymes we are actually investigating the structure of two proteases from different viral classes: human astrovirus, belonging to Picornaviruses and Rabbit hemorrhagic disease virus which belongs to Caliciviruses. The astrovirus protease is a serine protease with a trypsin-like fold; we solved the crystal structure of this enzyme to 2.0 Å resolution, revealing some unexpected features: i) a peculiar conformation of the catalytic Asp-His-Ser triad in which the aspartate side-chain is oriented away from the histidine, being replaced by a water molecule; ii) an unusual conformation and composition of the S1 pocket; and iii) the lack of the typical picornavirus surface β-ribbons together with a “featureless” shape of the substrate-binding site. The Rabbit hemorrhagic disease virus is a 3C-like cysteine protease; despite several efforts we did not manage to crystallize it, therefore we are now exploring different approaches to encourage crystallization: use of different constructs, surface mutagenesis and cocrystallization with substrate peptides. 33 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.07 CORONAVIRUS NONSTRUCTURAL PROTEIN 16 IS A CAP-0 BINDING ENZYME POSSESSING (NUCLEOSIDE-2’O)-METHYLTRANSFERASE ACTIVITY Etienne Decroly, Isabelle Imbert, Bruno Coutard, Mickaël BOUVET, Barbara Selisko, Karin Alvarez, Alexander E. Gorbalenya, Eric J. Snijder, and Bruno Canard Department of Structural Virology & Drug design AFMB/ESIL 163 Avenue de Luminy Case 925 13288 Marseille Cedex 09 France The coronavirus family of positive-strand RNA viruses includes important pathogens of livestock, companion animals, and humans, including the SARS-coronavirus that was responsible for a worldwide outbreak in 2003. The unusually complex coronavirus replicase/transcriptase is comprised of 15 or 16 virus-specific subunits that are autoproteolytically derived from two large polyproteins. In line with bioinformatics predictions, we now show that Feline coronavirus (FCoV) nonstructural protein (nsp) 16 possesses an AdoMet-dependent RNA (nucleoside2’O)-methyltransferase (2’O-MTase) activity that is capable of cap-1 formation. Purified recombinant FCoV nsp16 selectively binds to short capped RNAs. Remarkably, an N7-methyl guanosine cap (7MeGpppAC3-6) is a prerequisite for binding. HPLC analysis demonstrated that nsp16 mediates methyl transfer from AdoMet to the 2’O position of the first transcribed nucleotide, thus converting 7MeGpppAC 7MeGpppA 3-6 into 2’OMeC3-6. The characterization of eleven nsp16 mutants supported the previous identification of residues K45, D129, K169, and E202 as the putative K-D-K-E catalytic tetrad of the enzyme. Furthermore, residues Y29 and F173 of FCoV nsp16, which may be the functional counterparts of aromatic residues involved in substrate recognition by the vaccinia virus MTase VP39, were found to be essential for both substrate binding and 2’O-MTase activity. Finally, the weak inhibition profile of different AdoMet analogues indicates that nsp16 has evolved an atypical AdoMet binding site. Our results suggest that coronavirus mRNA may carry a cap-1 onto which 2'O methylation follows an order of events in which 2’O-methyl transfer must be preceded by guanine N7 methylation, with the latter step being performed by a yet unknown N7-specific MTase. 34 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.08 Computational Methods in Structural Virology for Investigating Drug Resistance, Viral Entry and Viral-Host Interactions Francisco S. DOMINGUES, Christoph Welsch, Oliver Sander, Hongbo Zhu, Ingolf Sommer, Thomas Lengauer Computational Biology and Applied Algorithmics Max-Planck-Institut Informatik Campus E1 4, Saarbrücken Germany We developed several structure-based computational approaches for investigating viral drug resistance, viral entry and viral-host protein interaction. We describe three medically relevant applications that involve HCV and HIV proteins. First, we have investigated the mechanisms of drug resistance to telaprevir in HCV[1]. Recent phase 1b clinical trial in patients infected with HCV genotype 1 revealed mutations in the NS3-4A protease that confer varying degrees of drug resistance. In particular, the mutated positions in two variants associated with drug resistance are located in the protein interior and not in the ligand binding pocket. Based on the available experimental structures of NS3-4A, we analysed the binding mode of different ligands and the network of non-covalent interactions within the protease structure to identify possible mechanisms of drug resistance. We then describe a method developed for predicting HIV-1 coreceptor usage upon cell entry[2]. HIV-1 cell entry commonly uses receptor CD4, as well as one of the chemokine receptors CCR5 or CXCR4 as coreceptor. Knowledge of coreceptor usage is critical for monitoring disease progression as well as for supporting therapy with the novel drug class of coreceptor antagonists. We show that using structural information on the V3 loop in combination with sequence features of V3 variants improves prediction of coreceptor usage. In particular, we propose a distance-based descriptor of the spatial arrangement of physico-chemical properties that increases discriminative performance. A detailed analysis and interpretation of structural features important for classification shows the relevance of several specific hydrogen-bond donor sites and aliphatic side chains to coreceptor specificity. Finally, we describe an approach for comparing protein-protein interactions that aligns vector representations of non-covalent interactions from different protein interfaces[3]. The method was applied to analyse the interaction between HIV gp120 and a CD4 mimic, demonstrating how this tool can assist in the development of mimetic antagonists. [1] Welsch C, Domingues FS, Susser S, Antes I, Hartmann C, Mayr G, Schlicker A, Sarrazin C, Albrecht M, Zeuzem S, Lengauer T. Molecular basis of telaprevir resistance due to V36 and T54 mutations in the NS3-4A protease of the hepatitis C virus. Genome Biol. 2008 9:R16. [2] Sander O, Sing T, Sommer I, Low AJ, Cheung PK, Harrigan PR, Lengauer T, Domingues FS. Structural descriptors of gp120 V3 loop for the prediction of HIV-1 coreceptor usage. PLoS Comput Biol. 2007 3:e58. [3] Zhu H, Sommer I, Lengauer T, Domingues FS. Alignment of non-covalent interactions at proteinprotein interfaces. PLoS ONE. 2008 3:e1926. 35 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.09 BENZIMIDAZOLES: POTENT FLAVIVIRIDAE INHIBITORS TARGETED AT THE RDRP IBBA C., Casula L., Collu D., Loddo R., Busonera B., Giliberti G., Iuliano F., Shukla S., Asthana S., PEZZULLO M. Department of Biomedical Science and Technology Section of General Microbiology and Virology & Microbial Biotechnologies University of Cagliari Italy We here report the highly potent and selective antiviral activity of four benzimidaziole derivatives (I, II, III, IV). In cell-based assays the above compounds showed EC50s in the range 0.8-1.5 μM against Bovine Viral Diarrhea Virus (BVDV), whereas they were inactive against Yellow Fever, Dengue and other positive-sense RNA viruses (such as poliovirus 1 and Coxsackie B2), negative-sense RNA viruses (Respiratory Syncyctial and Vesicular Stomatitis) and a double-stranded RNA virus representative (Reo 1). To get some insights into the target of the above compounds, drug-resistant viruses were selected by serial passages of wild type BVDV in the presence of stepwise doubling drug concentrations, starting from cell cultures infected with an m.o.i. of 0.01 and treated with drug concentrations equal to the EC50s. Sequence analysis of resistant cDNAs revealed the presence of single aminoacid substitutions in the coding region of the RNA-dependent RNA polymerase (RdRp). I selected for the I261M mutation; II selected for the N264D mutation; III selected for both I261M and N264D mutations; IV selected for the A392E mutation. Target validation was obtained with a newly developed fluorescence-based RdRp assay, in which all benzimidazoles inhibited the BVDV enzyme in a dose-dependent manner. They also inhibited the HCV1b RdRp, although with much lower potency. Automated Docking and Molecular Dynamics methods were used to help us to clarify issues related to binding site and binding modes of the most potent benzimidazoles, thus allowing to identify features undetectable by other means. Molecular modelling studies revealed that all the above NNI mutations are located in the fingers domain of the BVDV RdRp, thus suggesting that, in this virus, the NNI binding site is present in a domain different from that (thumb) reported for the same class of NNIs in HCV. 36 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.10 USING REPLICATIVE INFIDELITY TO TEST VIRAL PROTEASES M. KURZ, J. Zhu, N. Stefan, C. Mandl and T. Skern Department of Medical Biochemistry Max F. Perutz Laboratories Dr.-Bohr-Gasse 9/3, A-1030 Vienna Austria Tick-borne encephalitis virus (TBEV), a member of the flavivirus family, is endemic in large parts of Europe as well as in parts of Russia, China and Japan. It causes an acute infection of the nervous system. Cases of TBE do still occur in Central Europe, despite the availability of an effective vaccine. TBEV has a single stranded positive sense RNA genome. The genomic information is translated into a polyprotein which is then cleaved into the mature proteins by both viral and cellular proteases. We want to establish a TBEV-replicon system, in which the formation of viral particles depends on only one specific cleavage event of a heterologously expressed foreign proteinase. Due to the error prone nature of the viral RNA-polymerase, random mutations are introduced into this proteinase during replication. Proteinase mutants that still have cleavage activity can be distinguished from inactive ones by the formation of virus like particles. Therefore, this system can be used to study structure-function relationships and inhibitor resistance. We constructed a modular proteinase containing both the relevant parts of the cofactor NS2B and the viral proteinase NS3, connected by a flexible linker and provided with a His-tag for purification. The substrate C-prM was translated in vitro utilizing rabbit reticulocyte lysate and 35S labeled Methionine. It provides a natural cleavage site for the NS2B3 and was used to check the activity of the recombinant protease. We successfully expressed and purified NS2B3. An inactive form with a mutation in the active site, NS2B3 S192A, is used for crystallization assays which are still ongoing. The active enzyme cleaves its natural substrate C-prM. To make our replicon system work, we need a substrate that is cleaved by the heterologously expressed proteinase but not by NS2B3. Therefore, the natural TBEV cleavage site was substituted by various cleavage sites of the HIV1 proteinase. Whether these substrates meets this demand is currently under investigation. 37 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.11 ARRANGEMENTS FOR THE REGULATED PROCESSING OF ALPHAVIRAL NONSTRUCTURAL POLYPROTEIN Aleksei LULLA1 and Andrey Golubtsov2 1Institute 2Institute of Technology, University of Tartu, Nooruse 1, 50411 Tartu, Estonia. of Biotechnology,P.O. Box 56 (Viikinkaari 9), University of Helsinki, 00014 Helsinki, Finland. Semliki Forest virus (SFV) is a type member of Alphavirus genus, which produces its replicase proteins in a form of ns-polyprotein precursor P1234 and their maturation is performed in a temporally controlled manner by papain-like protease activity of nsP2 protein. A large body of evidence suggests that template preference and enzymatic capabilities of alphaviral replication complex have very important connection with its composition irreversibly altered by proteolysis. Final cleavage within polyprotein at the late stages of infection leading to maturation of nsP2 and nsP3 proteins is believed to denote the “point of no return” for viral replication, since plus-strands are no more accepted as templates and this puts emphasis on virions production in place of replicase self-amplification. Numerous previous studies devised rules for when and how ns-protease acts. Nevertheless, the understanding of the molecular bases for these processing rules and the triggers of specific cleavage of particular site at the right moment of replication remained rather illusive. The data obtained in our study aimed at investigating the molecular details of the regulation of the processing of 2/3 site in SFV ns-polyprotein provided evidence that socalled macro-domain (170 aa) of nsP3 protein is used for precise positioning of a substrate recognition sequence at the catalytic center of the protease and this process is coordinated by exact amino-terminal end of the N-terminal domain of nsP2 protein, thus representing the unique regulation mechanism used by alphaviruses. Although maturation of alphaviral replicase was always considered to be an attractive model for studies of fine mechanisms of proteolytic processing, it can now be concluded that even in such a seemingly simple system the efficiencies and the character of the cleavages performed by the same protease are obviously different and all three sequentially organized processing events are regulated by use of different mechanisms, only some of which are common for both most studied alphaviruses, SFV and Sindbis virus. Experimental data also disclosed the involvement of viral RNA in the regulation of the processing of 2/3 site suggesting the multimeric nature of the alphaviral replication complex residing on a viral RNA in a specifically ordered manner. Based on a newly available data a model for replicase complex formation was proposed, which may apply for replicases of several positive-strand RNA viruses. 38 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.12 STRUCTURAL AND FUNCTIONAL STUDIES OF FLAVIVIRAL PROTEINS NS3 AND NS5. Eloise MASTRANGELO#, Mario Milani#, Michela Bollati#, Graziella Sorrentino# and Martino Bolognesi# #Dept. of Biomolecular Sciences and Biotechnology University of Milan Via Celoria 26, 20133-Milan, Italy The development of new antiviral drugs is a priority in view of the worldwide viral threats to human health. In the context of the EC Integrated Project “Vizier” we are characterizing the Flavivirus replicative machinery through the study of the Viral Replication Complex. In particular we have performed structural and functional studies of the NS3 and NS5 proteins that are the main actors of viral replication as components of a multiprotein complex. During the replication, the viral genome is transcribed into a negativestrand RNA, used as template to synthesise the daughter viral genomic RNA. Besides the evident role of the RNA-dependent RNA polymerase (RdRp, NS5 Cterminal domain, Malet et al., 2007), to maintain proper viral replication the nascent transcript must be unwound from its complementary template RNA. Such role is performed by the C-terminal domain of NS3 (helicase domain). We analysed different NS3 constructs showing that full length NS3 displays increased helicase activity. Such results suggest that the protease domain (i.e. NS3 N-terminal domain) plays an assisting role in the RNA unwinding process. We investigated the interaction between the helicase and protease domains in NS3 by small angle X-ray scattering, producing a low resolution model showing scarce interaction between the two domains. These results suggest that domain rearrangements affecting the protease/helicase contact interface may occur in the RNA-bound protein (Mastrangelo et al., 2007). Once synthesized, RNA must be properly capped (cap I structure). This process requires almost three enzymes: a RNA-triphosphatase (again NS3 C-terminal domain), a guanylyltransferase and a N7 and 2'O MTase (the NS5 N-terminal domain). We recently demonstrated a possible involvement of NS5 also during the second step of RNA capping process. In fact, N-terminal domain of NS5 can bind covalently GMP. Such reaction may be the first step in the achievement of the 5' 5' bond between RNA diphosphate and GMP during the guanylyltransferase reaction (Bollati et al., in preparation). Following the characterization of single components of the viral replication machinery, we performed several experiments (pull-down, cross-linking, coexpressions) to obtain part of the poliprotein complex, involving the helicase and the RdRp domains, however without noticeable success so far. - Malet H, Egloff MP, Selisko B, Butcher RE, Wright PJ, Roberts M, Gruez A, Sulzenbacher G, Vonrhein C, Bricogne G, Mackenzie JM, Khromykh AA, Davidson AD, Canard B (2007). J Biol Chem. 14: 10678-89. - Mastrangelo E, Milani M, Bollati M, Selisko B, Peyrane F, Pandini V, Sorrentino G, Canard B, Konarev PV, Svergun DI, de Lamballerie X, Coutard B, Khromykh AA, Bolognesi M. (2007) J. Mol. Biol. 372, 444-55. 39 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.13 ACTIVE SITE JUMPING IN TAILSPIKE PROTEINS OF THE PODOVIRIDAE FAMILY OF TEMPERATE BACTERIOPHAGES AND PHAGE HEAD-BINDING MIMICRY IN CRYSTALS J. J. MÜLLER*, S. Barbirz§, A. Seul§, R. Seckler§, U. Heinemann* *Max-Delbrück-Centrum für Moleculare Medizin, 13125 Berlin, Robert-Rössle-Straße 10, Germany §Physikalische Biochemie, Universität Potsdam, 14476 Golm, Karl-LiebknechtStraße 24-25, Germany Sf6, HK620, and P22 belong to the Podoviridae family of bacteriophages that infect gram-negative bacteria by insertion of their double-stranded DNA. With their up to six tailspike proteins (TSP) attached to the phage head, they recognize and cleave their host cell receptor, i.e. the O-antigen. The trimeric TSP have highly homologous N-terminal head binding domains but no homology in their receptor binding parts, designated TSPN. We have determined the crystal structures of the TSPN of Shigella flexneri phage Sf6 to 1.25 Å (1), of Escherichia coli phage HK620 to 1.38 Å (2), and the first complete P22 tailspike structure to 1.65 Å resolution. All three tailspikes show a conserved architecture of the receptor binding part, i.e. a central, right-handed helix, flanked N-terminally by a mostly -helical region, and C-terminally by diverse β folds. The Sf6 TSP C-terminal domain consists of a β sandwich reminiscent of viral capsid proteins. Complex structures of the TSPs with their O-antigen fragments and corresponding biochemical analysis show a Shigella cell wall O-antigen fragment to bind to an endorhamnosidase active site located between two β-helix subunits each anchoring one catalytic carboxylate. The functionally and structurally related bacteriophage P22 TSP (3) and HK620 TSP have their active sites on single subunits. Sf6 TSP may serve as an example for the evolution of different host specificities on a similar general architecture. By introduction of the point mutation Y108W into the P22 TSP, the structure of a complete tailspike consisting of head binding domain, neck, and receptor binding part has been determined for the first time. With the help of trimeric structures of some additional chimeric constructs, made of the tailspike head binding domain and a long α helix, we could explain the mechanics of the neck kinking during biological processes which has been predicted from cryo-EM data. By moderate mechanical stress, the tryptophans Y108 will be drawn out of their hydrophobic pockets positioned between two subunits. Thereby, the chains are extended by about 5Å, which renders possible a kink within the head binding domain and receptor domain connecting neck. Crystallographic contacts provide forces which mimic the kinking and bend the neck by about 6°, thereby destroying the crystallographic threefold symmetry found so far in all TSPN trimers. (1) (2) (3) Müller, J.J., Barbirz, S., Heinle, K. Freiberg, A., Seckler, R., Heinemann, U. (2008) Structure, 16, 766-775. Barbirz, S., Müller, J.J, Uetrecht, C., Clark, A.J., Heinemann, U., Seckler, R. (2008) Mol. Microbiol., in press. Steinbacher, S., Baxa, U., Miller, S., Weintraub, A., Seckler, R., Huber, R. (1996). Proc. Natl. Acad. Sci. USA 93, 10584-10588. 40 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.14 RESIDUE L143 OF THE FOOT-AND-MOUTH DISEASE VIRUS LEADER PROTEINASE IS A DETERMINANT OF CLEAVAGE SPECIFICITY David NEUBAUER, Christina Mayer, Aloysius T. Nchinda, Regina Cencic, Katja Trompf and Tim Skern Department of Medical Biochemistry Max F. Perutz Laboratories Dr. Bohrgasse 9/3, A-1030 Vienna Austria The foot-and-mouth disease virus (FMDV) leader protease (Lpro) is a specific papainlike cysteine proteinase for which only three substrates have been as yet identified. These are the viral polyprotein which is cleaved by the proteinase in an intramolecular self-processing reaction and the two homologues of eukaryotic initiation factor 4G (eIF4G). The self-processing reaction at the Lpro/VP4 junction (LysLeuLys*GlyAlaGly) is severely impaired by replacing leucine at P2 with phenylalanine. Molecular modeling and energy minimization identified the L pro residue L143 as being responsible for this discrimination. The mutation L143A in L pro was sufficient to restore the efficiency of the self-processing reaction to wild type levels on a substrate with P2 leucine whereas the mutation L178A was not. The mutant L143A also showed improved self-processing on the sequence AspPheGly*ArgGlnThr present on eIF4GII. Only leucine and methionine can be found at position 143 of all sequenced FMDV serotypes. The fact that the variant L143M also showed delayed self-processing on the cleavage sites containing phenylalanine at P2 implies that these two bulky side chains are a determinant of the restricted specificity of Lpro. 41 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.15 THE ROLE(S) OF GENOMIC RNA IN MS2 CAPSID ASSEMBLY Óttar ROLFSSON and Peter Stockley Astbury Centre for Structural Molecular Biology University of Leeds Leeds, LS2 9JT United Kingdom Viral capsids are composed of protein subunits that in the complex environment of the cell are able to selectively package their genome and self-assemble into a structure of defined shape and size. How viruses achieve this is a major problem within structural virology. The possible role of the genome in this process is poorly understood. We are focusing on the role(s) of the viral genome in ssRNA virus capsid assembly employing the phage MS2 as our model system. MS2 is a bacteriophage comprised of a 3569 nt ssRNA genome protected by a T = 3 icosahedral capsid and a single maturation protein necessary for cell entry. The phage capsid itself is composed of 180 coat protein subunits. Our goal is to define the contributing effect of genomic RNA in MS2 capsid assembly. Our approach involves monitoring the efficiency of capsid assembly using gel shift assays, EM and sedimentation velocity analysis by comparing capsid assembly reactions in which different segments of the MS2 genome have been used to initiate capsid assembly. These investigations suggest that the coat protein subunits act to fold the MS2 RNA into the volume of the viral capsid and that RNA sequence/structure is therefore an important factor for efficient capsid assembly. 42 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.16 INVESTIGATING THE INFLUENCE OF DIMERISATION ON THE ACTIVITY OF THE LEADER PROTEASE OF FOOT AND MOUTH DISEASE VIRUS Jutta STEINBERGER and Tim Skern Department of Medical Biochemistry Max F. Perutz Laboratories 1030 Vienna, Dr.-Bohr-Gasse 9/3 Austria The leader protease (Lbpro) of foot and mouth disease virus frees itself from the growing polypeptide chain during translation by cleavage between its own Cterminus and the N-terminus of the subsequent protein VP4. Previous crystallography and NMR studies revealed that the C-terminal extension (CTE) of Lbpro is bound by the substrate-binding site of a neighbouring molecule resulting in the formation of a stable dimer in solution. The amino acid residues W105 and T117, located at the interface between the globular domains of the two Lbpro molecules, are potentially involved in dimer formation. Therefore, we used PCR mutagenesis to substitute these residues with alanine. Both single mutations as well as the double mutation W105A T117A were fully active in self-processing. Nevertheless, the mutation W105A was not sufficient to disturb dimer formation, as assayed by size exclusion chromatography. Therefore, we tried to provoke repulsion between dimerized Lb pro molecules by substituting W105 with the positively charged amino acid arginine. However, Lb pro bearing the W105R mutation showed full activity in self-processing. Analysis of dimer formation of this and other mutated proteins will be presented. Supported by the Austrian Science Foundation. 43 | P a g e VIZIER / SPINE2 Workshop on Structural Virology P.17 THE ROLE OF SPECIFIC HISTIDINES AS PH SENSORS IN FLAVIVIRUS MEMBRANE FUSION Karin STIASNY, Richard FRITZ, and Franz X. HEINZ Institute of Virology Medical University of Vienna 1095 Vienna, Kinderspitalgasse 15 Austria Flaviviruses enter cells by receptor-mediated endocytosis and fusion of the viral with the endosomal membrane. The fusion event is triggered by the acidic pH of this compartment and is mediated by the major envelope protein E, a class II viral fusion protein. At acidic pH the native metastable E dimers dissociate leading to the exposure of a previously buried functional element (fusion peptide, FP) that initiates fusion by interacting with the target membrane. Further conformational changes result in the generation of a trimeric postfusion structure. Several histidines are absolutely conserved in the E protein of all flaviviruses and because of their pKa have been hypothesized to function as molecular switches for fusion. To provide experimental evidence for the role of these residues in flavivirus fusion, we studied the effect of their replacement on membrane fusion in the context of recombinant subviral particles of tick-borne encephalitis virus. Surprisingly, replacement of three of the absolutely conserved histidines had virtually no effect on fusion-related processes. However, mutating a histidine (H323) located at the interface between domains I and III led to a strong impairment of E dimer dissociation, FP exposure, and binding to target membranes; i.e. the early stages of fusion. These results suggest that histidine 323 forms part of the low-pH trigger required for initiating flavivirus membrane fusion. 44 | P a g e VIZIER / SPINE2 Workshop on Structural Virology 45 | P a g e VIZIER / SPINE2 Workshop on Structural Virology LIST OF ALL PARTICIPANTS Carme ARNAN Structural Biology IBMB-CSIC C/Baldiri Reixac 10-12 I-20133 Barcelona, Spain carcri@ibmb.csic.es Dennis BAMFORD Instiute of Biotechnology University of Helsinki PL 56 (Viikinkaari 5) 00014 Helsinki, Finland dennis.bamford@helsinki.fi Mads BEICH-FRANDSEN Dept. for Biomolecular Structural Chemistry University of Vienna Campus-Vienna-Biocenter 5 1030 Vienna, Austria mads.beich-frandsen@univie.ac.at Dieter BLAAS Dept. for Medical Biochemistry Medical University of Vienna Dr.-Bohr-Gasse 9/3 1030 Vienna, Austria dieter.blaas@meduniwien.ac.at Martino BOLOGNESI Dept. for Biomolecular.Sciences & Biotechnology University of Milano Via Celoria, 26 I-20133 Milano, Italy martino.bolognesi@unimi.it Daniele BONIVENTO Dept. for Genetics and Microbiology University of Pavia Via Ferrata 1 27100 Pavia, Italy daniele.bonivento@uniroma1.it Mickael BOUVET AFMB UMR6098 University of Aix-Marseille I & II ESIL/AFMB Case 925 13288 Marseille Cedex 09, France mickael.bouvet@afmb.univ-mrs.fr Gerard BRICOGNE Global Phasing Ltd. Sheraton House Castle Park CB3 0AX Cambridge, United Kingdom Gb10@globalphasing.com Bruno CANARD AFMB University of Méditerranée Marseille Campus de Luminy 13288 Marseille, France bruno.canard@afmb.univ-mrs.fr Siva CHARAN DEVANABOY Dept. for Structural Biology University of Graz Humboldstr. 50/3 8010 Graz, Austria siva.charan@uni-graz.at Lionel COSTENARO Structural & Computational Biology Institute for Reseachr in Biomedicine Parc Científic de Barcelona 08028 Barcelona, Spain lionel.costenaro@irbbarcelona.org Stephen CUSACK EMBL Grenoble Outstation 6 rue Jules Horowitz 38042 Grenoble, France cusack@embl.fr Kristina DJINOVIC-CARUGO Dept. for Biomolecular Structural Chemistry University of Vienna Campus Vienna Biocenter 5 1030 Vienna, Austria kristina.djinovic@univie.ac.at Francisco DOMINGUES Computational Biology & Applied Algorithmics Max-Planck-Institut for Informatics Campus E1 4 66123 Saarbrücken, Germany doming@mpi-sb.mpg.de Aleksandra FLANAGAN Division of Structural Biology University of Oxford Welcome Trust Centre, Roosevelt Drive OX3 7BN Oxford, United Kingdom Aleksandra@strubi.ox.ac.uk Claus FLENSBURG Global Phasing Ltd. Sheraton House Castle Park CB3 0AX Cambridge, United Kingdom claus@globalphasing.com 46 | P a g e VIZIER / SPINE2 Workshop on Structural Virology Richard FRITZ Institute of Virology Medical University of Vienna Kinderspitalgasse 15 1095 Vienna, Austria richard.fritz@meduniwien.ac.at Stephen GRAHAM Division of Structural Biology University of Oxford Welcome Trust Centre, Roosevelt Drive OX3 7BN Oxford, United Kingdom stepheng@strubi.ox.ac.uk Jonathan GRIMES WTCHG University of Oxford Welcome Trust Centre, Roosevelt Drive OX3 7BN Oxford, United Kingdom jonathan@strubi.ox.ac.uk Rolf HILGENFELD Institute of Biochemistry University of Lübeck Ratzeburger Allee 160 23538 Lübeck, Germany hilgenfeld@biochem.uni-luebeck.de Eric G. HUIZINGA Crystal & Structural Chemistry University of Utrecht Padualaan 8 3584-CH Utrecht, The Netherlands e.g.huizinga@uu.nl Cristina IBBA Dept. for Biomedical Science & Technology University of Cagliari Sesta Strada Ovest 09010 Macchiareddu (Ca), Italy cristinaibba@microbiologia.ca.it Anna JANSSON Dept. for Cell & Molecular/Structural Biology University of Uppsala Husargatan 3 75124 Uppsala, Sweden anna.jansson@icm.uu.se Jan KADLEC EMBL Grenoble Outstation BP 181, 6 rue Jules Horowitz 38042 Grenoble Cedex 9, France kadlec@embl.fr Martina KURZ Dept. for Medical Biochemistry Medical University of Vienna Dr.-Bohr-Gasse 9/3 1030 Vienna, Austria martina.kurz@meduniwien.ac.at Hongmin LI New York State Department of Health Wadsworth Center 120 New Scotland Ave. 12208 Albany, USA lih@wadsworth.org Aleksei LULLA Institute of Technology University of Tartu Nooruse 1 504111 Tartu, Estonia lulla@ut.ee Katsumi MAENAKA Medical Institute of Bioregulation University of Kyushu 3-1-1 Maidashi, Higashi-ku 812-8582 Fukuoka, Japan kmaenaka@bioreg.kyushu-u.ac.jp Ioannis MANOLARIDIS EMBL Hamburg Outstation Notkestrasse 85 22603 Hamburg, Germany manolari@embl-hamburg.de Eloise MASTRANGELO Science Biomolecular & Biotechnology University of Milano Via Celoria, 26 20133 Milano, Italy Eloise.mastrangelo@mi.infm.it Ulrike MAURER Molecular Structural Biology Max-Planck-Institute of Biochemistry Am Klopferspitz 18 82152 Martinsried, Germany maurer@biochem.mpg.de Mario MILANI Science Biomolecular & Biotechnology University of Milano Via Celoria, 26 20133 Milano, Italy mario.milani@mi.infm.it Jürgen MÜLLER Crystallography Max-Delbrück-Center Robert-Rössle-Strasse 10 13125 Berlin, Germany jjm@mdc-berlin.de David NEUBAUER Dept. for Medical Biochemistry Medical University of Vienna Dr.-Bohr-Gasse 9/3 1030 Vienna, Austria david.neubauer@univie.ac.at 47 | P a g e VIZIER / SPINE2 Workshop on Structural Virology Jan PAESHUYSE Rega-Institute Virology & Exp. Chemotherapy K.U. Leuven Minderbroedersstraat 10 3000 Leuven, Belgium jan.paeshuyse@rega.kuleuven.be Karen PANGERL Institute of Virology Medical University of Vienna Kinderspitalgasse 15 1095 Vienna, Austria karen.pangerl@meduniwien.ac.at Magnus PERSSON Dept. for Cell & Molecular/Structural Biology University of Uppsala Husargatan 3 75124 Uppsala, Sweden magnusj@xray.bmc.uu.se Margherita PEZZULLO Dept. for Biomedical Science & Technology University of Cagliari Sesta strada Ovest, Z.I. di Macchiareddu 09010 Uta (CA), Italy margheritapezzullo@microbiologia.ca.it Félix REY Dept. for Structural Virology Instiute Pasteur 25 rue de Dr. Roux 75015 Paris, France rey@pasteur.fr Ottar ROLFSSON Astbury Centre for Structural Molecular Biology University of Leeds Manton building, Clarendon Way LS2 9JT Leeds, United Kingdom bmbor@leeds.ac.uk Rob RUIGROK Unit of Virus Host Cell Interactions University Joseph Fourier 6 rue Horowitz, B.P. 181 38042 Grenoble Cedex 9, France ruigrok@embl.fr Oliver SANDER AG3 Max-Planck-Institute for Informatics Campus E 1 4 66123 Saarbrücken, Germany osander@mpi-sb.mpg.de Eike SCHULZ Dept. for Molecular Structural Biology University of Göttingen Justus-von-Liebig-Weg 11 37077 Göttingen, Germany eschulz1@gwdg.de Oliver SMART Global Phasing Ltd. Sheraton House Castle Park CB3 0AX Cambridge, United Kingdom osmart@globalphasing.com Vernon SMITH Bruker AXS B.V. Technical Sales Manager Structural Biology Oostsingel 209 2612 HL Delft, The Netherlands vernon.smitz@brucker-axs.nl Jutta STEINBERGER Dept. for Medical Biochemistry Medical University of Vienna Dr.-Bohr-Gasse 9/3 1030 Vienna, Austria jutta.steinberger@univie.ac.at Alasdair STEVEN Laboratory of Structural Biology Research National Institutes of Health 50 South Drive, room 1517 20892 Bethesda, Maryland, USA dq1n@nih.gov Karin STIASNY Institute of Virology Medical University of Vienna Kinderspitalgasse 15 1095 Vienna, Austria Karin.stiasny@meduniwien.ac.at Paul TUCKER EMBL Hamburg Outstation Notkestrasse 85 22603 Hamburg, Germany tucker@embl-hamburg.de Mark VAN RAAIJ Dept. for Structural Biology Instituto de Biologia Molecular de Barcelona c/Josep Samitier 1-5 08028 Barcelona, Spain vanraaij@usc.es Herman VAN-TILBEURGH IBBMC University of Paris Sud Bat 430 91405 Orsay, France herman.van-tilbeurgh@u-psud.fr Winfried WEISSENHORN UVHCI c/o CIBB University Jospeh Fourier 6 rue Jules Horowitz 38042 Grenoble, France weissenhorn@embl.fr 48 | P a g e VIZIER / SPINE2 Workshop on Structural Virology Justyna WOJDYLA EMBL Hamburg Outstation Notkestrasse 85 22603 Hamburg, Germany tyyna@embl-hamburg.de Thomas WOMACK Global Phasing Ltd. Sheraton House Castle Park CB3 0AX Cambridge, United Kingdom twomack@globalphasing.com Jürgen ZLATKOVIC Clinical Institute of Virology Medical University of Vienna Kinderspitalgasse 15 1095 Vienna, Austria juergen.zlatkovic@meduniwien.ac.at 49 | P a g e VIZIER / SPINE2 Workshop on Structural Virology Venue Institute of Molecular Biotechnology, IMBA Dr.-Bohr-Gasse 3, A-1030 Vienna IMBA is in the building number 2 on the map. The venue will take place in the main lecture hall of IMBA. 50 | P a g e VIZIER / SPINE2 Workshop on Structural Virology Travel Directions IMBA is located in the 3rd district (3. Bezirk; postal code 1030) of Vienna. Public Transport Stations close to the MFPL/IMBA: Railway: S7 St. Marx (100m away) Tram: 71 or 18 “St. Marx” (right in front) or 18 “Viehmarktgasse” Bus: 74A “St. Marx” (right in front) Underground: U3 “Schlachthausgasse” (additional 7 min walk or two stations with tram 18 to “Viehmarktgasse”) From the Vienna International Airport Wien Schwechat: Railway S7 directly to St. Marx/Vienna Biocenter City Airport Train (CAT) to “Wien Mitte”, additionally bus 74A to “St.Marx” Vienna Airport Shuttle Bus to the train station “Südbahnhof”, additionally tram 18 to “St. Marx” Taxi Car: motorway A4, Exit Knoten Prater From the train stations: Südbahnhof: tram 18 to “St.Marx” Westbahnhof: tram 18 to “St.Marx” Franz-Josefs-Bahnhof: U4 to “Landstraße” (= “Wien Mitte”), additionally railway S7 or bus 74A to “St.Marx” Arriving by Car: A4: Exit Knoten Prater A23: Exit Erdberg Lectures will take place in the lecture hall of Institute of Molecular Biotechnology (IMBA) at Campus Vienna Biocenter, Dr. Bohrgasse 3, 1030 Vienna. The Campus Vienna Biocenter is Austria's largest life-sciencies park, which encompasses Research Institute of Molecular Pathology (IMP), Institute of Molecular Biotechnology (IMBA), Gregor Mendel Institute of Molecular Plant Biology (GMI) and the Max F. Perutz Laboratories (MFPL) of the University of Vienna and of Medical University. 51 | P a g e VIZIER / SPINE2 Workshop on Structural Virology Where to eat in Vienna’s third District Here just a few of the numerous restaurant suggestions: Irodion Landstraßer Hauptstraße 71, 1030 Wien Greek, daily 11:30 – 24:00 Credit cards will be accepted Lubin Hainburger Straße 48, 1030 Wien Mail: reservierung@lubin.at; www.lubin.at Fish restaurant; daily 11.00-15.00 and 17.30-24.00 Credit cards will be accepted Restaurant im Hotel Artis Rennweg 51, 1030 Wien Mail: hotel@artis.at www.artis.at Mon-Sat 11.00-22.00, warm meals 11.00-14.00 and 18.00-22.00 Credit cards will be accepted Salm Bräu Rennweg 8, 1030 Wien Mail: office@salmbraeu.com Viennese and international specialities Mon-Sun 11.00-24.00 Visa accepted Akula 1030 Wien, Ungargasse 21 - 23 Indish; daily 11.00-14.30 and00-23.00 CC: Amex, Diners, Mastercard, VISA, JCB Amon’s Gastwirtschaft 1030 Wien, Schlachthausgasse 13 E-Mail: office@amon.at Mon-Sat 10.00-24.00, Sun 10.00-16.00 CC: Amex, Diners, Mastercard, VISA, Hobex Asia Restaurant Lou 1030 Wien, Landstraßer Hauptstraße 139 Asian; daily 11.30-15.00 and 17.00-22.30 CC: VISA, Mastercard Akakiko 1030 Wien, Landstraßer Hauptstraße 59 E-Mail: info@akakiko.at Japanese; daily 10.30-24.00 Bar-Celona 1030 Wien, Rasumofskygasse 34 E-Mail: bar-celona@gmx.at Spanish; Mon-Sat 9.00-22.00 Credit cards will not be accepted. Bieramt 1030 Wien, Am Heumarkt 3 E-Mail: bieramt@bieramt.at Pub; daily 11.00-1.00. Kitchen until 24.00 CC: Amex, Diners, Mastercard, VISA Burgenland Vinothek 1030 Wien, Baumannstraße 3 E-Mail: office@burgenland-vinothek.at Vinotheken / Weinbar; Tue-Fri 13.0019.00, Sat 10.00-17.00 CC: VISA, Mastercard, Bankomat Asia Running Sushi Paradies 1030 Wien, Landstraßer Hauptstraße 99 Telefon: 01/7151494 Japanese; Mon-Fri 10.00-20.00, Sat 10.00-18.00 Credit cards will not be accepted. Klabautermann 1030 Wien, Markhofgasse 4 Mon-Fri 9.00-02.00, Sat & Sun 17.00-24.00 Credit cards will not be accepted. Modena 1030 Wien, Landstraßer Hauptstraße 133 E-Mail: pizzeria_modena@chello.at Italian Restaurant; Mon-Fri 11.00-24.00, Sat 12.00-4.00, Sun 12.00-22.30 CC: VISA, Mastercard, Bankomat www.pizzeriamodena.at 52 | P a g e VIZIER / SPINE2 Workshop on Structural Virology Conference dinner will take place in “Wiener Rathauskeller” Wiener Rathauskeller Rathausplatz 1 A-1010 Wien Tel.: +43 (1) 405 1210 Fax: +43 (1) 405 1219-27 e-mail: office@wiener-Rathauskeller.at It can be reached from the venue location by: - tram 71 from St Marx to Schwarzenberg Platz and then change to - tram D from Schwarzenberg Platz to Rathaus (We will hand out your tram tickets at registration.) 53 | P a g e VIZIER / SPINE2 Workshop on Structural Virology Wireless information These are the usernames and passwords to enter the WLAN-System at our workshop venue. Accounts are active from 14th July 2008 09:00 to 16th July 2008 17:00. Username Password imba/guest4 73354855 54 | P a g e guest5 35466725 guest6 03333768 guest8 11209378 guest9 87997864 guest10 70686201 guest11 89991773 guest12 15539269 guest13 23149593 guest14 37907579 guest15 96552483 guest16 86328437 guest17 21908131 guest18 86012123 guest19 36833535 guest20 69872184 VIZIER / SPINE2 Workshop on Structural Virology