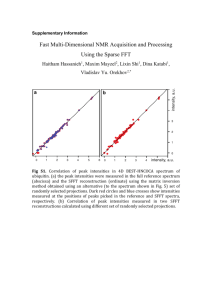
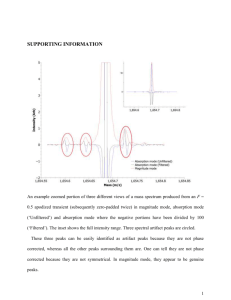
emi12565-sup-0003-si
advertisement

1. Supporting Materials and Methods Chemicals All chemicals were purchased in analytical grade from Sigma-Aldrich (Taufkirchen, Germany) and Merck (Darmstadt, Germany), respectively. Terminology Analytics: Throughout this text we use the term ‘peak’ to designate a mass spectral feature which is described by a mass-to-charge ratio (m/z) and a retention index (RI). The term ‘analyte’ designates a chromatographic peak which is the convolute of a single or many mass spectral features. For downstream analysis each analyte is represented by a single mass spectral feature using its peak height for analyte quantification. Datasets: The ‘metabolite dataset’ comprises the normalized measured peak heights (analyte abundances) for each evaluated peak across the metabolite profiles and contains missing values (Tab. S3). The ‘quantification dataset’ designates the metabolite dataset where the replicates of the replicate groups with completely missing values are replaced by random values sampled from a Gaussian distributed as simulated background / noise distribution. This dataset still contains missing values. The ‘complete dataset’ designates the quantification dataset with fully imputed missing values. GC/MS data processing and compound identification Peak alignment: An initial non-targeted metabolite fingerprint alignment was generated using msPeak to construct a targeted peak library for further quantitative analyses. For this, the GC/MS chromatograms were exported using Leco ChromaTOF software (version 3.25) as NetCDF files (Andi MS format) with the following settings: baseline subtraction just above the noise, peak width broadening using width-to-time (w/t) of 3.5 / 0 sec – 6.0 / 1 400 s. Peak smoothing (as running mean with 7 data points), peak apex search (as local maxima search with 11 data points) and alignment using peak spread and peak gap to neighboring peaks were performed within msPeak for each mass trace separately. The peak list for each chromatogram was exported for post-processing. Peak redundancy: To reduce the redundancy of peaks potentially derived from the same analyte, a time - similarity (T/S) clustering approach was used (Krueger et al 2011). In detail, peaks were grouped according to their RI (adjusted retention time based on FAME retention) into time groups with 0.25s resolution. The similarities among time group members were computed on raw peak heights using Pearson and Spearman correlation (Sokal 1995). Both similarity matrices were averaged (sØ) and converted into a distance range using the equation (1 – sØ) / 2. The distance matrices were then clustered using hierarchical average linkage clustering (Legendre 1998) and the resulting trees cut at 0.146 heights (sØ = 0.707). Thus, the obtained clusters reflect about 50% of the local variance among assigned peaks within a cluster. The T/S clusters were manually evaluated and curated (i) to remove ambiguous or badly shaped peaks, (ii) to identify a single peak as reliable quantification fragment for an analyte, and (iii) to remove already known analytical byproducts and artifacts. The resultant peak library consists of 643 quantification peaks including analytical standards and thus represents 4.5% of the initial peak set. This peak library was used for targeted peak extraction with narrow time windows (on average ±0.8s around the peak specific average RI) using the exported peak lists. Peak annotation: Compound identification was performed using several reference compound libraries of commercially available authentic standards measured as described (Huege et al 2011, Kopka et al 2005, Krall et al 2009). All annotations were manually curated and linked to the quantification peak-dataset. GC/MS data analyses Data normalization: In subsequent data analyses steps various profile-wise normalization approaches were applied. To keep the normalized peak heights within the original measurement scale, each sample profile was divided by the ratio of its corresponding profile-specific scale parameter to the mean of estimated scale parameters across all profiles. Data pre-processing: To account for analytical variations during sample derivatization and extraction the quantification peak-dataset was normalized first to the total ion count of the RI markers and secondly to the U-13C-Sorbitol peak height. All 14 analytical standards were afterwards removed. To further filter the dataset for relevant peaks, following criteria were applied: (i) peaks detected in at least 50% of the measured biological replicates (3 out of 5, majority rule), (ii) peaks clearly above their corresponding blank peaks, i.e. the average sample peak height minus 2 times standard error is larger than the corresponding blank average peak height plus 2 times standard error, (iii)peaks revealing an average fold change greater than the robust average fold change within the blank replicates with a lower and upper fold-change bound of 1.5 and 3, (iv) peaks revealing an average peak height of ≥1000 arbitrary units All criteria must be fulfilled in at least one out of the nine replicate groups (genotype × treatment) to consider a peak as being validly measured. This filtering resulted in 529 quantification peaks that are kept for further downstream analysis. Subsequently, the blank samples were removed resulting in a filtered data matrix of 529 peaks and 45 profiles. To account for sample amount variations, the profiles were normalized using the optical density as biomass equivalent. Data post-processing: To further correct for experimental and technical variations a modified version of the Progenesis CoMet normalization was applied (Nonlinear Dynamics Limited, 2014). This approach aims to identify robust, non-changing peaks considered as "spikes" for estimating a robust scaling factor. Briefly, peaks were expressed as log2-ratios to their corresponding groupwise reference profile, estimated using one-step Tukey's biweight (Affymetrix, 2002) over the five biological replicates. Outliers were removed globally using a full (n-step) Tukey's biweight until no further outlying ratios were detected. In total, 90 peaks were considered robust, as no outlying ratio was detectable, and thus, were used for profile normalization. The scaling factor for each profile was estimated and applied as the anti-log of the mean of the log-ratios. The resultant dataset was further normalized using variance-stabilizing transformation without calibrating (Huber et al 2002). Data evaluation: The resultant dataset was evaluated for univariate outliers over all peaks within each of the nine replicate groups. Only values detected by both, a boxplot statistics and one-step Tukey's biweight are considered as outliers. In total, 944 (3.97%) univariate outliers were detected and treated as missing values. Subsequently, the peaks were evaluated for reliable quantification using their groupwise coefficient of variation (CV). For this, the mean average, the Tukey's biweight, and the median over all groupwise CV values were estimated and outliers were removed by iterative boxplot statistics. Peaks revealing outlying values in the majority of cases (2 out of 3) were removed from further analysis. The final metabolite dataset (Tab S3) comprises 501 peaks and 45 sample profiles with a total of 2082 (9.23%) missing values. Data imputation: For 93 out of all 4509 combinations of peak and replicate group no measureable peak was detectable in the 5 biological replicates, illustrating qualitative differences between genotypes and / or treatments. These peaks are most likely below the detection level or within the noise of the method rather than truly absent. For those 93 combinations a simulation approach was chosen to simulate values within the methods background / noise for missing value imputation. To estimate global noise, the median and the 95% quantile were computed for combinations with ≤2 measurements (minority rule), resulting in 437 and 1419 arbitrary units respectively. The standard error (SE) was estimated as 95% quantile for all combinations with >3 measurements (majority rule) revealing a groupwise maximum peak height of less than 1419 arbitrary units. The resulting global noise was considered as 437 ± 212 (as mean and SE) and used to random generate values from a normal distribution constrained with the global noise parameter. The resultant quantification dataset contains 7.17% missing values. The remaining missing values were imputed by principal component analyses (PCA) on log2transformed and mean-centered data using Bayesian model, probabilistic model as well as by a linear combination of the k most significant eigengenes (Stacklies et al 2007) and the resulting complete datasets were back-transformed. The robust mean estimated using one-step Tukey’s biweight of the three different imputation outcomes was used to finally replace missing values. Statistical analyses and visualization All data and statistical analyses were performed if not otherwise stated according to (Sokal 1995) and (Legendre 1998) using R 3.0.2 (R Development Core Team, 2013). Univariate outliers, extreme deviations from the center, were detected by (i) boxplot statistics and (ii) Tukey's biweight. The boxplot statistics was performed with a coefficient of 1.5 and values which lie beyond the extremes of the whiskers were considered as outliers (cf. (Steinhauser et al 2011)). The Tukey's biweight algorithm was performed with a tuning constant of 5 (Affymetrix, 2002) and observations with a weight of 0.0 were considered as outliers. Both approach were executed either in one step or iteratively with n-steps, i.e. repeated until no outliers were detectable anymore. Multivariate outliers were detected by calculating the robust Mahalanobis distance based on a robust estimate of the center and the covariance with the minimum covariance determinant (MCD) estimator (Rousseeuw and Van Driessen 1999). Outliers were detected by using the 97.5% quantile of a chi-square distribution with p degrees of freedom (χ2p), as the observed distances are approximately chi-square distributed. Coefficients of variation (CV), the ratio of the standard deviation to the mean, were expressed as percentages. The uniqueness of peaks was estimated using a time similarity clustering approach (see above) with varying time window and similarity cutoffs. The uniqueness was computed as the fraction of clusters with one member to the total of estimated clusters and expressed as percentage. Euclidean distances were computed either on the log2-transformed datasets or on the estimated component scores from principal component analyses and subjected to various clustering and visualization approaches. Cluster trees were drawn on the basis of hierarchical cluster analysis (HCA) using average linkage clustering. Partitioning around medoids (PAM, k-medoids), a more robust version of k-means clustering, was used for clustering sample profiles (Kaufman 2005). K-means clustering was performed for finding peaks revealing similar abundance patterns using initialization with cluster centers derived from HCA analysis. Gap statistic, a goodness of clustering measure, was performed to estimate the number of supported clusters (Tibshirani et al 2001). Euclidean distance matrices were converted into principal coordinates space using classical multidimensional scaling (CMD; (Cox 2000)). Non-parametric analysis of variance by means of Mantels test was performed as Pearson’s matrix correlation (rm) with 9999 bootstrap samples between the Euclidean distance matrix and a design matrix, a binary (0, 1) matrix representing cluster assignments. The silhouette information (SI), a combined measure of the separation among and cohesion within cluster / cluster members, was expressed as mean silhouette width (Rousseeuw 1987). Both cluster validity indices yield values in the interval of [-1, 1], in which larger positive values reflect more favorable cluster solutions. The variation of information (VI) index was used to evaluate the similarity between observed and expected clustering’s (Meila 2007) with VI values closer to 0 revealing a better agreement. Principal component analysis (PCA) was performed on the log2-transformed and mean-centered complete dataset using singular value decomposition (Venables 2002). The number of optimal principal components (PC’s) were estimated using parallel analysis and optimal coordinates approach (cf. (Cangelosi and Goriely 2007)). Confidence ellipses were drawn for a 95% region using the correlation, mean and standard deviation of data points. Two-way factorial analysis of variance (ANOVA) was performed with genotype, treatment (time), and genotype × treatment interaction as factors. Tukey's ‘Honest Significant Difference’ (HSD) method was used as post-hoc test to estimate the differences between the means of the levels of factors. If not otherwise stated all p-values (p) were corrected by Benjamini – Yekutieli correction to control the false discovery rate in multiple testing (Benjamini and Yekutieli 2001).