Treatment - Angelfire
advertisement

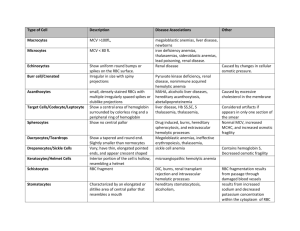
Nelson’s 14th Addition Chapter16 (pp.1232-1256) Anemia’s Anemia results when RBC’s can’t be produced in sufficient numbers to replace those removed from circulation. 1. Congenital Pure RBC Anemia (ex. Congenital Hypoplastic Anemia) rare condition usually seen in early infancy characteristic diagnostic feature is deficiency of RBC precursor in normal bone marrow genetic basis with male = female some pt.’s with tryptophan metabolism abnormality ADA (adenosine deaminase) activity increased in RBC Increased erythropoietin in serum and urine Manifestations Pale appearance (50%) Profound anemia 2-6 months Heart failure and death without transfusion Macrocytic normochromic anemia Assay reveals young erythrocyte population Thrombocytosis and occasional neutropenia Despite increased erythropoietin there is lack of erythropoietic activity Diminished reticulocytes Treatment Corticosteroids Transfusion is the only chance for survival if corticosteroids do not produce remission 10-15% do not respond to corticosteroids and require transfusion every 4-6 weeks 2. Acquired Pure RBC Anemia’s Cause unknown Removal of thymus tumor have led to remissions in some Associated in thymoma in children Erythropoietin inhibiting antibody, antibodies to erythroblasts and inhibitors of heme synthesis have been identified in plasma May respond to corticosteroids Immunosuppressive's may be given if corticosteroids ineffective Markedly reduced erythrocyte precursors in marrow Decreased RBC survival rate is basis of aplastic crisis in some hemolytic anemia’s 3. Anemia’s of Chronic Infection, Inflammation and Renal Disease Anemia complicates many systemic diseases (ex. Chronic pyogenic infections) Important symptoms and signs underlie disease Normochromic normocytic anemia with low serum iron and elevated serum ferritin (acute phase reactant) If underlying systemic disease is controlled, anemia spontaneously remises 4. Physiologic Anemia of Infancy Progressive decline of hemoglobin levels within the 1st week of life which persists for 6-8 weeks Several factors are operative 1. abrupt cessation of erythroporesis 2. shortened RBC survival 3. expansion of blood volume with rapid weight gain (1st 3 months) pre term infants have greater decline in hemoglobin concentrations dietary factors may aggravate anemia ( high portions of polyunsaturated fatty acids with iron supplements) vitamin E deficiency presenting with “infantile pyknocytosis” (self limited hemolytic process, large numbers of acanthocytes) diet with essential nutrients for hemopoietic process 5. Megaloblastic Anemia’s RBC’s are larger than normal in every stage Finely dispersed chromatin in nucleus Asynchrony between cell cytoplasm and nuclear maturity Increased RNA in proportion to DNA Uncommon in US A. Folic Acid Deficiencies Caused by deficient folic acid due to decreased intake/absorption Rapid growth or infection compounds deficiency Human and cow milk have adequate folic acid Manifestations Low birth weight Peak incidence 4 – 7 months Irritability, fail to gain weight and chronic diarrhea Thrombocytopenic hemorrhages in advanced cases May accompany Kwashiorkor or Marasmiro Progressive anemia RBC count disproportionately lower than hematocrit RBC shape and size variations Low reticulocyte count Elevated serum activity of LDH (lactate dehydroginase) Treatment Parenteral folic acid 2-5 mg/24 hr. x 3-4 weeks Transfusion with severe anemia Folic acid as low as 50 g/24 hr. can be used to test between primary folic acid deficiency and B12. 1. Megaloblastic Anemia of Pregnancy Increased folate demand 1 mg/24 hr. folate supplement 2. Folic Acid (FA) Deficiency of Malabsorption FA absorbed through small intestine Celiac disease, chronic infectious enteritis or any other inflammatory or degenerative disease of intestine can interfere with FA absorption Oral FA 1mg/24 hr. is treatment 3. Congenital Defect of FA Absorption Defect in intestinal absorption of FA Inability to transfer folate from plasma to CNS Oral folic acid 15-50mg/24 hr. 4. FA Deficiency complicating Hemolytic Anemia’s Chronic hemolysis may increase FA requirement Adults>children Transfusion may be needed Examine bone marrow if anemia worsens or there is an increased need for transfusion Normal diets may eliminate need for FA supplement 5. FA Deficiency associated with Anticonvulsant's and Other Drugs Anticonvulsant's may lead to decreased FA Usually no anemia or symptoms Possible displacement of FA from serum carrier FA therapy B. Vitamin B12 Deficiencies Intrinsic factor needed for B12 absorption Absorbed in terminal ileum Dietary deficiency rare Seen in Vegans (no milk, eggs or animal products) Most cases due to absorption problem Manifestations Juvenile onset 9 months to 10 years Irritability, anorexia and listlessness Painful smooth red tongue Neurological symptoms: ataxia, paresthesia, hyporeflexia, + babinski’s, clonus and coma Serum iron and FA may be normal or elevated Moderate bilirubin levels Excessive excretion of methylmalonic acid in urine is reliable for diagnosis B12 absorption assessed by schilling test Treatment Parenteral B12 1 – 5 g/24 hr. Maintenance for life Oral therapy contraindicated 1. Transcobalamine Deficiency Transcobalamines I and II are plasma B12 binding proteins with type II being primary Autosomal recessive Severe megaloblastic anemia in early infancy Massive parenteral B12 therapy 2. Vitamin B12 Deficiency in Older Children Atrophy of gastric mucosa and achlorhydria possible Cutaneous candidiosis and thyroid/endocrine deficiencies possible Parenteral B12 regularly Structurally abnormal intrinsic factor seen 6. Microcytic Anemia’s A. Iron Deficiency Anemia Lack of iron for hemoglobin synthesis Most common hemotologic disease in infancy and childhood 8 – 15 mg iron in diet is necessary newborns iron mostly in circulating hemoglobin usually during 1st 4 – 6 months due to dietary insufficiency blood loss must be considered in every case ⅓ infants with severe iron deficiency in US have chronic intestinal loss induced by exposure to a heat labile protein in whole cow’s milk Manifestations Pallor most important clue Few symptoms may be seen Spleen enlarged and palpable Obese or underweight children Pica may be prominent May have effects on neurological and intellectual function Serum ferritin is relatively accurate to iron stores RBC’s become smaller with less hemoglobin Reticulocyte count is normal Treatment Oral administration of ferrous salts Parenteral therapy in calculated doses Diet considerations B. Sideroblastic Anemia Heterogeneous Abnormal iron or heme metabolism Ringed marrow sideroblasts found X linked form appears in childhood 7. Hemolytic Anemia’s Shortened RBC survival Increased marrow activity Increased reticulocyte count X-ray changes seen in skull and fingers Elevated unconjugated bilirubin Plasma hemoglobin increases Isotopic techniques can estimate RBC survival 8. Intrinsic Abnormalities of the RBC A. Hereditary Spherocytosis No abnormalities of hemoglobin except glucose-6-phosphate dehydrogenase (G-6-PD) type Most common hereditary hemolytic anemia Most common among Northern Europeans Autosomal dominant – occasionally recessive Abnormality of spectrin (protein lattice of RBC’s shape and lipid bilayer) Spleen involves-hemolytic process ceases after removal Onset in infancy 50% of unsplenectomized patients form gallstones reticulocytosis, anemia and hyperbilirubinemia spherocytic RBC’s smaller than normal osmotic fragility studies indicated Treatment splenectomy (over 5 years old) – eliminates aplastic crisis and chance of gallstones 9. Enzymatic Defects of RBC’s A. Pyruvate Kinase Deficiency Congenital – autosomal recessive (homozygous) Impaired ATP, Pyruvate and NAD Increased 2,3 DPG Reduced RBC life span Jaundice and anemia in neonatal period Spiculated pyknocytes present Diagnosis relies on reduced pyruvate kinase activity Treatment Exchange transfusion Packed RBC transfusion Splenectomy 10.Glucose-6-Phosphate Dehydrogenase Deficiency Inherited More common in blacks A. Drug Induced Hemolytic Anemia Males > Females No G-6-PD deficiency evident Antipyretics, sulfonamides, antimalarials and fava beans(favism) Death may occur Premature black infants can have spontaneous hemolysis Heinz Bodies seen (only 1st 3-4 days of onset) Diagnosis based on decreased G-6-PD activity in RBC’s 11.Hemoglobin Disorders A. Sickle Cell Severe chronic hemolytic disease Premature destruction of RBC’s RBC’s brittle Ischemic changes may also be involved No characteristics may be present Ischemic necrosis of small bones Acute painful vaso-occlusive episodes most frequent and prominent Ischemic damage may also affect myocardium, liver and kidney’s Splenomegally enlargement possible Treatment Maintain full immunization status Prophylactic PCN-G Oral acetaminophen for painful episodes or codeine if needed B. Thalassemia Syndromes Heterogeneous and hypochromic Decreased or absence of mRNA for 1 or more globin chains Many mutations can occur Most prevalent of all genetic diseases Most prevalent regions are those associated with malaria from Plasmodium Falciparum 1. Thalassemia (Thalassemia Major AKA Cooley’s Anemia) 2-6 months of life Regular transfusions necessary Thin bones which can lead to pathologic fractures Enlarged liver and spleen Diabetes Mellitus can occur Fragmented poikilocytes and target cells present Nucleated RBC’s Elevated unconjugated bilirubin Treatment Transfusions -Hemosiderosis is inevitable due to prolonged transfusion therapy *Thalassemia major is AKA Cooley’s Anemia and associated with facial deformities such as severe maxillary hyperplasia and malocclusion. 2. Thalassemia Deficient globulin chain(s) Most severe form results from deletion of all 4 globin chains 12.Extrinsic Factors A. Autoimmune Hemolytic Anemia’s(with “warm” antibodies) Uncertain pathology Possibly drugs or infectious agents Acute onset Pallor, jaundice, pyrexia and hemoglobinemia Splenomegally Response to corticosteroids and full recovery within 3 months are characteristic of acute form Other form is chronic + direct coomb’s test Treatment Transfusions Prednisone 2-5 mg/kg/24 hr. Remits spontaneously in weeks to months B. Autoimmune Hemolytic Anemia’s(with “cold” antibodies) 1. Cold Agglutinin Disease Severe episodes of intravascular hemolysis with hemoglobinemia and hemoglobinuria may occur 2. Paroxysmal Cold Hemoglobinuria Specific cold antibody – Donath-Landsteiner Hemolysin ⅓ cases associated with congenital or acquired syphilis transfusions for severe anemia avoid chilling patient 13.Intoxication’s and Infections Arsenic and phenylhydrazine produce hemolysis Hemolytic anemia’s may complicate various infections Septicemia may cause direct RBC damage Parasites can affect RBC’s