Adipokines: Regulators of Metabolic Integration and Energy Metabolism
advertisement

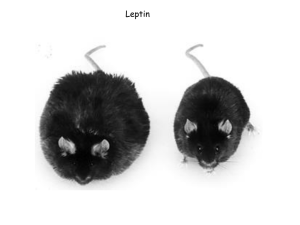
Adipokines: Regulators of Metabolic Integration and Energy Metabolism Tsu-Shuen Tsao Christopher Hug Harvey F. Lodish As the primary energy reservoir, adipose tissue has long been recognized as an important player in the regulation and maintenance of energy homeostasis. Expansion of adipose tissue leads to obesity, which is associated with a number of non–adipose-centered conditions such as type 2 diabetes mellitus, hypertriglyceridemia, dyslipoproteinemia, hypertension, and coronary artery disease (1,2,3). Nevertheless, adipose tissue had been viewed more as a storage structure for fat than as a regulator like pancreatic β-cells. With the discovery of adipose-derived factors and recognition of their regulatory roles in homeostatic maintenance, adipose tissue has emerged as a major endocrine organ (4,5). Factors secreted from adipose tissue participate in many physiologic processes: body weight and appetite regulation, lipid and carbohydrate metabolism, immunity and inflammatory response, vasculature and blood pressure maintenance, and reproduction. The importance of adipose tissue in metabolic regulation has been demonstrated by the abnormalities observed in humans with congenital generalized lipodystrophy (6) and in lipotrophic mice (7,8). Consequences of lacking adipose tissue include insulin resistance, hyperglycemia, hypertriglyceridemia, and fatty liver (6,7,8). Although loss of triglyceride storage sites can explain some of these defects, namely hypertriglyceridemia and fatty liver, it cannot account for insulin resistance and diabetes. These conditions are most likely due to loss of regulatory factors secreted from adipose tissue. These important regulatory factors have come to be known as adipokines. The term is derived from cytokine, which in current usage loosely defines a molecule secreted by a cell that affects its own behavior or that of another cell. Therefore, in this chapter the term adipokine is used to describe a cytokine produced from adipose tissue. It is synonymous with adipocytokine, which is also commonly used. In addition to adipocytes, adipokines can also be produced from other cell types that compose adipose tissue fibroblasts, immune cells, and cells forming the blood vessels (5). We think the term adipokine should be reserved specifically for factors secreted from adipocytes. It should be emphasized that there are two main adipose tissue depots: visceral and subcutaneous. These two depots differ metabolically (9,10). Visceral or central obesity, characterized by elevated waist to hip ratio, is more closely associated with insulin resistance (9,10). Expression and secretion of many adipokines described below differ between visceral and subcutaneous depots (11,12). For some adipokines, the contribution from one depot to circulating levels outweighs the other (4,11). For this reason, conclusions based on comparison of adipokine level in one specific depot versus that in serum must be drawn carefully. Where appropriate, we will make an effort to specify if secretion of a specific adipokine is increased in one depot versus the other. Leptin The idea that circulating factors can regulate food intake is not new. As early as the 1950s, when the circulatory system of a normal lean rat was surgically fused to that of a rat made obese by ventromedial hypothalamus lesion, its appetite decreased and became thinner (13). This work and subsequent parabiosis studies using the diabetic db/db mice suggested the presence of a satiety factor that was overproduced by the unresponsive obese rodents (14). When responsive rodents were exposed to an excess of this satiety factor through the parabiotic exchange, they became anorectic. The search for this satiety factor ended with the positional cloning of the mouse obese, or ob, locus (15). First reported in 1950 (16), homozygous mutation at this locus in mice resulted in a myriad of metabolic abnormalities: hyperphagia, hyperglycemia, hyperinsulinemia accompanied by pancreatic β-cell dysfunction, increased metabolic efficiency, thermogenesis defects, massive fat depot, and impaired fertility (14,17). Those ob/ob mice in parabiosis with either wild-type lean mice or db/db mice were no longer hyperphagic and lost weight (18). These studies predicted that the ob locus is responsible for generating a circulating anorectic factor. Indeed, the recombinant product of the ob locus reduced food intake and body fat mass while it increased energy expenditure in both ob/ob and wild-type mice (19). For this reason the protein encoded by the ob gene is called leptin, derived from leptos, meaning “thin” in Greek. P.964 To date, leptin is the best characterized adipokine. The complementary DNA for leptin predicts an 18-kd protein (15). Following cleavage of the signal peptide, the mature leptin is a 16-kd circulating protein secreted mainly from adipocytes (15). Although low levels of leptin expression have been found in stomach, placenta, mammary gland, skeletal muscle, and brain, they do not contribute significantly to the circulating leptin pool (4,20). Circulating leptin levels are closely correlated with body fat content and adipocyte size (21,22,23,24,25). The molecular mechanisms responsible for this phenomenon remain unclear. Unlike insulin, another metabolically important molecule, intracellular trafficking and secretion of leptin is not acutely regulated (26). However, its expression is influenced by many factors. Insulin, glucocorticoids, increased body weight due to overfeeding or high-fat feeding, tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), and bacterial endotoxin all can elevate leptin expression as well as circulating leptin levels, whereas fasting, testosterone, thiazolidinedione treatment, β-adrenergic agonists/catecholamines, and exposure to cold have the opposite effect (20). The nervous system may also play a role in regulation of leptin production. Adipose tissue is innervated by the sympathetic nervous system (27,28). Some evidence suggests that leptin expression can be reduced by the sympathetic nervous system via the β3adrenoceptor present on adipocytes (4). Because leptin can stimulate autonomic nervous activity (described below), inhibition by the sympathetic nervous system may provide a negative feedback loop to control circulating leptin levels. As the early parabiosis studies might predict, the receptor for leptin is encoded by the db locus (29). Leptin receptor (LR, or ObR) has many splice variants (30). All share the same extracellular domain, but only the b splice variant (LRb) contains a long cytoplasmic domain capable of signal transduction (30). LRb is highly expressed in the hypothalamus (30,31), the area critical for feeding regulation. A mutation leading to altered splicing and truncation of the LRb cytoplasmic domain is responsible for the obese and diabetic phenotypes of the db/db mice (29,30). Some of the short splice variants still contain a portion of the cytoplasmic domain present in LRb. It is unclear whether these variants can transduce signal from the remaining cytoplasmic regions. Two of the recognized physiologic functions of the short splice variants are transported through the blood–brain barrier and clearance from circulation (31). Structural studies have revealed that leptin folds into a four-alpha-helix bundle structure characteristic of the hematopoietic cytokine superfamily that includes erythropoietin, growth hormone, IL-2, and prolactin (32,33). It is thus not surprising that LRb belongs to the hematopoietin/cytokine receptor family that includes the erythropoietin receptor and the common gp130 subunit of IL-6 and leukemia inhibitory factor receptors (34). Just like other members of this superfamily, binding of LRb to leptin activates signal transducers and activators of transcription (Stat) family of transcription factors via Janus protein tyrosine kinases (Jak) (35,36). LRb lacks an intrinsic catalytic activity. To transduce its signal, the binding of leptin to its receptor allows the associated Jak2 to transphosphorylate itself, leading to its activation and subsequent tyrosine phosphorylation of LRb (36,37). LRb contains three cytoplasmic tyrosine residues. Phosphorylation of Tyr1138 permits transcription factor Stat3 to dock [through its src homology 2 (SH2) domain], where it is subsequently phosphorylated by Jak2 (36,37,38,39,40). In tissue culture systems leptin receptor can activate many different Stat isoforms (39,41), but so far only activation of Stat3 has been shown in the hypothalamus (42). Stat3 is not the only signaling pathway affected by leptin. Leptin has been shown to stimulate phosphatidylinositol 3-kinase (PI 3-kinase) (43,44,45,46) and mitogen-activated protein kinase (MAPK, or ERK, for extracellular signal-regulated kinase) (36,40,45,47). Activation of MAPK requires binding of protein tyrosine phosphatase SHP-2 to Tyr985 of LRb (40,48). The contributions of these signaling pathways to the many physiologic effects of leptin have not been quantitatively analyzed. Whether there is cooperativity among the different signaling pathways also remain to be worked out. Major physiologic functions of leptin include appetite regulation, energy balance, and sexual maturation and reproduction. Both the autonomic nervous system and peripheral tissues are direct targets of leptin action. In addition, leptin can have indirect effects on peripheral tissues through the sympathetic nervous system. LRb is expressed in discrete regions of the brain and at high levels in the ventrobasal and ventromedial hypothalamus (49). Mice receiving leptin via peripheral injection, or more potently via direct infusion into cerebral ventricles, lost weight by eating less and expending more energy (50). The central effects of leptin are orchestrated by a battery of neuropeptides, some of which are produced by neurons that express LRb (51). Because the intricacies of neuroanatomy underlying the functional basis of these neuropeptides cannot be adequately described here, readers should refer to reviews devoted to this subject (49,52,53). In simplistic terms, leptin directly or indirectly regulates expression of two opposing groups of neuropeptides: orexigenic and anorexigenic. Orexigenic neuropeptides include neuropeptide Y (NPY), agouti-related peptide (AgRP), melanin-concentrating hormone (MCH), and orexin (54). Anorexigenic peptides include corticotropin-releasing hormone (CRH), cocaine- and amphetamine-regulated transcript (CART), and a cleaved product of proopiomelanocortin (POMC), α-melanocyte stimulating hormone (α-MSH) (54). In general, leptin decreases the expression of orexigenic neuropeptides while it stimulates the expression of anorexigenic peptides. These neuropeptides in turn influence food intake as well as energy balance (49). Neurons expressing NPY, POMC, AgRP, and CART also express LRb and are thought to be direct targets of leptin, whereas the others are affected secondarily through neuronal projections from the direct targets (54). Leptin-sensing neurons in the hypothalamus also extend leptin’s action into the pituitary and the adrenal glands. Hypercorticosteronemia is a hallmark of adrenal abnormality in ob/ob mice and can be suppressed by leptin treatment before significant weight loss occurs (14,49). Decreased leptin levels may account for the elevated glucocorticoid levels and suppression of the immune system during starvation (4,20). Another neuroendocrine effect of leptin mediated through the hypothalamus–pituitary axis is stimulation of thyroid hormone release (20,54). This may constitute a large part of leptin’s role in energy balance P.965 regulation because thyroid hormones stimulate thermogenesis and basal metabolic expenditure. Leptin has functions other than feeding behavior and energy balance regulation. Leptin is required for female mice to reach puberty and sexual maturity (55). Female ob/ob mice are infertile even when body weight was normalized by caloric restriction (55). This reproductive defect can be corrected by administration of leptin (56). Finally, leptin can affect glucose and lipid metabolism in skeletal muscle and liver and insulin secretion from pancreatic β-cells (20). The metabolic effects of leptin observed in mice suggest that it signals satiety and is an antiobesity hormone. However, leptin may not be a satiety signal in all species. Some studies suggest that it can regulate short-term feeding in rodents but not in humans (54). However, in both humans and rodents, obesity is characterized by high leptin levels (22,24). If leptin’s role is to promote satiety and prevent obesity, it is an open question why obesity is characterized by high leptin levels. Several lines of evidence suggest that leptin resistance may explain the inability of high leptin levels to prevent obesity. Leptin is thought to enter the nervous system through a truncated splice variant of LR, LRa, that lacks much of the LRb intracellular domain and is present at high levels in brain microvessels (57). The ratio of leptin levels in cerebrospinal fluid versus plasma is reduced in obese individuals (58). The response of some obese mice to leptin was higher when delivered directly into the nervous system than peripherally (50). This suggests that delivery of leptin to its site of action in the central nervous system (CNS) is saturated in obese animals. Another component of leptin resistance may be inhibition of leptin receptor signaling. One transcriptional target of Stat3 following activation by leptin is suppressor of cytokine signaling-3 (SOCS-3) (59). SOCS-3 inhibits Stat3 activation by binding to phosphorylated Tyr985 of leptin receptor, not by competitive binding to Tyr1138 (60). Protein tyrosine phosphatase 1B (PTP1B) can inhibit leptin receptor signaling by dephosphorylating Jak2 (61,62). In addition, mice deficient in PTP1B are hypersensitive to insulin and leptin, and resist weight gain (61,62). Is there a physiologic role for leptin that is consistent with its myriad metabolic effects? The existence of leptin resistance indicates that leptin is a poor antiobesity hormone. In addition, the effectiveness of leptin treatment on normalizing energy balance in obese humans is limited (63). In humans as well as in mice, leptin levels decrease acutely following food withdrawal (64,65). In the starvation state, lack of leptin signals danger and reduces energy consumption by decreasing energy expenditure, diminishing immune responses, and shutting down reproduction. When starvation is no longer a danger, sufficiency of plasma leptin permits more energy-requiring physiologic processes to take place. Thus, current data may be more compatible with leptin as a signal for nonstarvation (20). Adipocyte Complement Related Protein of 30 kd (Acrp30)/Adiponectin Adipocyte Complement Related Protein of 30 kd (Acrp30) was first identified as a protein expressed and secreted by differentiated murine 3T3-L1 adipocytes (66). It was independently cloned as AdipoQ (67), gelatin-binding protein of 28 kd (GBP28) (68), and adiponectin (69,70). It is an abundant serum protein (66). Acrp30 has four readily recognized domains: a 17– (mouse) or 18–amino acid (human) residue N-terminal signal sequence, a conserved 17–amino acid region, a collagen domain with 21 perfect Gly-X-Pro or Gly-X-Y repeats, and a C-terminal globular domain with sequence homology to C1q. Acrp30 is highly conserved among mammalian species (71). It is part of a family of proteins that contain sequences homologous to the C1q globular domain (72,73). Some members of this family also contain a collagen domain, although the number of collagen repeats differs greatly (72). The x-ray diffraction pattern of the Acrp30 globular domain (aa 111–247) crystals revealed a homotrimeric structure with a central hydrophobic core formed by the interface of the three individual monomers (74). The 10-strand jelly roll topology showed Acrp30 to be a structural homologue of the TNF-α family of trimeric cytokines even though they share little primary sequence homology. Analysis of human and mouse plasma by size-exclusion gel chromatography or sucrose velocity gradient centrifugation revealed that Acrp30 forms multiple species of different apparent molecular weights (66,68,75). High-resolution size-exclusion chromatography showed that Acrp30 in mouse serum exists as three separate species with apparent molecular weights of approximately 630,410, and 210 kd (76). The same species were found in recombinantly produced Acrp30 (76). Equilibrium sedimentation using the analytic ultracentrifuge identified the 410- and 210-kd species as hexamers and trimers of Acrp30 with actual molecular weights of 143 and 80 kd, respectively (76). These differences are likely due to the collagen domains, because they are known to adopt an extended conformation that can exaggerate the molecular weight of a protein in size-exclusion chromatography. Whereas the hexamer and the 630-kd high-molecular-weight (HMW) isoforms of Acrp30 comprise the majority of serum Acrp30, Acrp30 trimer is the predominant isoform secreted by differentiated 3T3-L1 adipocytes (76). Three major lines of evidence implicate Acrp30 in regulation of energy metabolism: altered expression and circulating levels in abnormal metabolic states, physiologic effects of recombinantly produced Acrp30, and genetic studies in humans and mice. The insulin resistance–inducing cytokine TNF-α reduced expression of Acrp30 in differentiated murine 3T3-L1 and primary human adipocytes (77,78,79). Expression of Acrp30 was found to be decreased in leptin-deficient ob/ob mice (67) and leptin receptor–deficient db/db mice fed a high-fat diet (80). The plasma concentration of Acrp30 is reduced in obese or diabetic humans (69,81,82,83) as well as in rhesus monkeys during the early stages of progression toward insulin resistance (84). Circulating Acrp30 concentrations were negatively correlated with human plasma triglyceride and fasting insulin levels (81,82). A longitudinal study of Pima Indians showed that those with high Acrp30 levels were less likely to develop type 2 diabetes mellitus (85). Expression of Acrp30 is reduced in healthy first-degree relatives of patients with type 2 diabetes (86), suggesting Acrp30 as a potential genetic determinant of insulin sensitivity. Acrp30 levels increase following weight loss (81,87), P.966 caloric restriction (88), cold exposure (75), and treatment with insulin-sensitizing peroxisome proliferator–activated receptor-γ (PPAR-γ) agonist thiazolidinediones (83,88,89). These data suggest that Acrp30 is an adipocyte-derived hormone that mediates the effects of thiazolidinedione drugs on restoration of insulin sensitivity and normal glucose homeostasis. Acrp30 administration elicits several metabolic changes in mice. Injection of the globular head region of Acrp30 (gAcrp30) accelerated removal of free fatty acids (FFAs) from circulation in mice, at least partly by increased muscle fatty acid oxidation (90). This effect was accompanied by a decrease in plasma glucose level independent of insulin and glucagon. In addition, chronic gACRP30 treatment resulted in reduction of body weight independent of food intake. An Acrp30 fragment containing the globular head could be found in human plasma, although its identity remains to be determined (90). Long-term treatment (12 days) with Acrp30, and more potently with gAcrp30, of mice improved insulin sensitivity, decreased storage of triglycerides in liver and muscle, and increased expression of proteins in muscle involved in fatty acid combustion (80). In addition, hyperglycemia, elevated plasma FFA levels, and increased muscle triglyceride content in insulin-resistant lipoatrophic, db/db, and yellow agouti KKAy mice were markedly improved upon Acrp30 treatment and even more so with very low doses of gAcrp30 (80). Lastly, Acrp30, but not gAcrp30, can increase the ability of subphysiologic levels of insulin (35 pM) to suppress gluconeogenesis in isolated hepatocytes (88). Infusion of Acrp30 inhibited hepatic glucose production in mice by decreasing the expression of gluconeogenic enzymes phosphoenolpyruvate carboxykinase and glucose-6-phosphatase (91). The different effects of full-length Acrp30 and gAcrp30 on muscle fatty acid oxidation and hepatic glucose production suggest that they may have different physiologic functions. In addition, there are several functional studies implicating Acrp30 in antiatherogenic and antiinflammatory processes (92,93,94,95,96). These functions of Acrp30 are mediated partly by antagonizing the proinflammatory effects of TNF-α on endothelial cells and macrophages (92,93,96) and partly by reducing cholesteryl ester and lipid accumulation in macrophages (94). Figure 65.1. Signaling by different isoforms of Acrp30. Both globular and full-length signal through AdipoR1 and AdipoR2 as described in the text. Higher-order forms of full-length Acrp30, depicted here as hexamer and high-molecular weight (HMW), stimulate nuclear factor κB through a hypothetical receptor, here represented by a question mark. FAO, fatty acid oxidation. Stimulation of muscle fatty acid oxidation by gAcrp30 is mediated at least in part by 5′adenosine monophosphate– activated protein kinase (AMPK) (97,98). AMPK is stimulated by increased cellular AMP/adenosine triphosphate (ATP) ratios and upstream kinases (99). AMPK plays a major role in stimulation of muscle fatty acid oxidation and glucose transport following exercise and treatment with metformin or the AMP analogue 5-amino 4imidazolecarboxamide riboside (AICAR) (100,101,102). The activity and phosphorylation of AMPK was found to be increased in isolated muscles incubated with gAcrp30 or muscles of mice directly injected with gAcrp30 (97,98). This was associated with phosphorylation and inactivation of the AMPK target protein acetyl-CoA carboxylase (ACC), leading to decreased levels of malonyl-CoA, an allosteric inhibitor of carnitine palmitoyltransferase and a key inhibitor of mitochondrial fatty-acid oxidation (97,98,99,103). Full length Acrp30 trimer can activate AMPK similar to gAcrp30, whereas full-length hexamer cannot (98). In contrast, hexameric and HMW Acrp30 isoforms can activate transcription factor nuclear factor κB (NFκB) in cultured C2C12 myoblasts or myotubes (76). The physiologic significance of this activity remains to be clarified. The current understanding of Acrp30 signaling pathways is illustrated in Fig. 65.1. Recently Yamauchi et al. identified two homologous receptors that tentatively can mediate the stimulatory effects of Acrp30 on fatty acid oxidation (104). Termed AdipoR1 and AdipoR2, both receptors bind gAcrp30 and full-length Acrp30, albeit with different affinities (104). Suppression of AdipoR1 or AdipoR2 expression in C2C12 myotubes by small interfering RNA (siRNA) reduced stimulation of fatty acid oxidation and glucose uptake by gAcrp30 and full-length Acrp30, respectively (104). Reduction of fatty acid oxidation and glucose uptake was more complete with decreased expression of both AdipoR1 and R2 (104). Increased AdipoR1 expression was also accompanied by increased phosphorylation of AMPK and ACC following gAcrp30 treatment in C2C12 myocytes and hepatocytes (104). AdipoR1 and R2 are predicted to contain seven transmembrane domains and are distant relatives of G protein– coupled receptor families (104). However, unlike all known G P.967 protein–coupled receptors, the N-termini of AdipoR1 and R2 face the cytoplasmic side, whereas their C-termini are exposed to the outside of the cell (104). In transiently transfected 293T cells, AdipoR1 can form heterodimers with AdipoR2 (104). Human genetic studies of Acrp30 have supported the results of these metabolic studies. Genome-wide scans of a large group of human subjects identified a quantitative trait locus on 3q27 that influences the phenotype of metabolic syndrome (insulin resistance, obesity, hypertension, and coronary artery disease) (105). One of the genes on 3q27 is Acrp30 (106,107). An independent study also mapped 3q27 as a locus for susceptibility of earlyonset diabetes in French whites (108). Several single nucleotide polymorphisms (SNPs) in the human Acrp30 locus itself are associated with low serum Acrp30 levels or metabolic syndrome components (109,110,111). It remains to be determined if these SNPs in the Acrp30 gene represent genuine mutations or merely are markers in linkage disequilibrium with SNPs in other genes that are the true regulators of Acrp30 levels and the metabolic syndrome. Metabolic studies in mice with a disrupted Acrp30 locus also indicate a role for Acrp30 in regulating energy metabolism. Acrp30-deficient mice exhibit delayed clearance of plasma FFAs, decreased muscle fatty acid transporter-1 (FATP-1) expression, and elevated adipose tissue and circulating TNF-α levels (112). Although Acrp30-deficient mice exhibited only a mild abnormality in glucose homeostasis when fed a standard rodent chow diet (113), marked insulin resistance developed with a high-fat diet (112). A third study of Acrp30-deficient mice did not find insulin resistance under regular or high-fat diets, but paradoxically showed increased β-oxidation in muscle and liver (114); the reasons for this disparity are unknown. Thus, a sizable body of literature has accumulated to support the view that Acrp30 regulates lipid and glucose metabolism hormonally and thus can play a critical role in the development of obesity and diabetes. However, much work is still needed to validate this hypothesis. First, although pharmacologic studies suggest that gAcrp30 is the active form for fatty acid oxidation and full-length Acrp30 is the active form for hepatic glucose production, the physiologically active forms of Acrp30 remain to be determined. Furthermore, different Acrp30 oligomers may have separate activities. Indeed, although hexameric and HMW Acrp30 isoforms are potent activators of the NFκB signaling pathway, the trimer form does so poorly (76). The significance of NFκB activation is currently unknown. Lastly, it will be important to identify the underlying causes responsible for the decline in Acrp30 levels associated with obesity and diabetes. Tumor Necrosis Factor-α TNF-α, first identified as a secreted molecule produced principally in the immune system, is a member of an increasingly large family of proteins (tumor necrosis factor superfamily, or TNFSF) that share a common type 2 transmembrane protein topology and a trimeric structure. Many members exist both as a transmembrane protein and as a cleaved, secreted protein. TNFSF members are capable of diverse actions ranging from inducing apoptosis to tissue organization (reviewed in reference 115). The conserved features of the TNFSF include a threefold axis of symmetry with a characteristic “jellyroll” β sandwich (115); in the case of TNF-α, the unprocessed membrane-associated molecule is 26 kd, whereas the proteolytically cleaved and secreted mature molecule is 17 kd. This processing is by the TNF-α–converting enzyme (TACE); also known as ADAM-17, it is a member of the ADAM (a disintegrin and metalloproteinase) family of proteases (116). Within the immune system, TNF-α has potent inflammatory effects, stimulating the secretion of cytokines (IL-6, among others) and leading to the coordinated response to infection of a variety of organisms. The action of TNF-α is mediated through the two TNF-α receptors, types 1 (p55 or p60) and 2 (p75 or p80), with the former more widely expressed. These receptors share homology in the extracellular domain important for ligand binding, but diverge substantially in the intracellular domains and in their pattern of expression. Although many of the TNFSF members act within the immune system, the range of cell types and tissues expressing TNF-α and its receptors is large and includes adipose tissue (117,118). The two receptors share different roles in mediating insulin resistance; soluble forms of the receptors exist as well, and the interplay between these molecules, serum TNF-α, and tissue TNF-α may ultimately determine the biologic effects of each cytokine. TNF-α is increased in a variety of systemic pathologic conditions, including inflammatory diseases such as rheumatoid arthritis, inflammatory bowel disease, and cardiac disease, and inducing elevated levels of TNF-α in animals has been shown to lead to a state of insulin resistance (119). The mechanism for this action is not completely understood, but involves both direct action of the hormone on target tissues and long-term metabolic compensatory changes accompanied by changes in gene expression. Secretion of TNF-α is required for the hyperglycemia and abnormal lipid profiles seen in obese mice, as evidenced by resistance of a TNF-α knockout mouse to the development of impaired insulin action (120). Adipocyte membrane–associated TNF-α that is incapable of being cleaved due to mutations in the TACE cleavage site can activate TNFR1 but not TNFR2 to exert local diabetic effects. TNF-α acts through pathways of transcription factors, including NFκB; these pathways are active in a variety of cell types and modulate many cellular physiologic pathways. Because many of these pathways converge on cellular metabolism, Ruan et al. (79) evaluated genes regulated by both short- and long-term TNF-α treatment. Short-term treatment of 3T3-L1 adipocytes (<24 hours) reduced messenger RNA (mRNA) for both Acrp30 and glucose transporter 4 (GLUT-4), with the effects greater with prolonged treatment; simultaneous treatment with insulin partially ameliorated this effect. The reduction at 24 hours of Acrp30 was 79%, and of GLUT-4 78%. In examining the global effects of TNF-α treatment on adipocytes, they found that the transcription of many were altered as early as 4 hours. Suppressed genes involved in mature adipocyte functions included transcription factors (C/EBP-α, PPAR-γ), metabolic pathways and enzymes (GLUT-4, glycogen synthase), and secreted molecules (Acrp30, angiotensinogen). Other genes, however, were induced, P.968 and many of these are expressed in preadipocytes; a subset of these, including plasminogen activator inhibitor-1 (PAI-1), secreted vascular cell adhesion molecule-1 (VCAM-1), fibronectin, tenascin, adrenomedullin, and ceruloplasmin are also overexpressed in type 2 diabetes. The targets of TNF-α are varied and include immediate [inhibition of insulin-induced insulin receptor and insulin receptor substrate 1 (IRS-1) phosphorylation (121,122,123)], short-term [increased insulin-induced phosphorylation of IRS-1 (124)], and long-term (suppression of adipocyte specific genes and induction of preadipocyte genes through an NFκB-dependent pathway). The components of the TNF-α signaling pathways are illustrated in Fig. 65.2. In order to examine the short- and long-term consequences of elevated TNF-α, Ruan et al. examined the global changes in gene expression in several tissues in rats infused with TNF-α for 1 or 4 days (125). Biochemical analysis revealed that basal glucose and C-reactive protein levels did not change significantly, but there were significant decreases in Acrp30 (50% decrease at both 1 and 4 days), elevated FFAs at 1 and 4 days, and increased triglycerides at 4 days. Despite the normal basal glucose levels, the animals became insulin resistant at both time points. These results are consistent with TNF-α exerting both immediate and long-term alterations in homeostasis; such changes might be expected to result from changes in gene expression. To examine altered gene expression induced by TNF-α, Ruan et al. (125) made use of DNA microarrays to examine the changes in gene expression in animals treated with the hormone for either 1 or 4 days. They observed only moderate alterations in muscle gene expression, with no change in genes known to be involved in glucose and fatty acid metabolism. In contrast, there were significant changes in adipose and liver gene expression, with more changes noted in the adipose tissue; potentially, both liver and muscle respond secondarily to changes conveyed through TNF-α actions on adipose tissue. In adipose tissue, TNF-α downregulated genes involved in the uptake and storage of both FFAs and glucose, and upregulated the expression of genes involved in the inflammatory response. Alterations in gene expression in the liver were again consistent with maintaining an elevated serum glucose (downregulated glycogen synthase 2 and GLUT-2) and elevated serum FFA levels (decreased expression of mitochondrial enzymes involved in β-oxidation). Figure 65.2. Signaling by tumor necrosis factor-α (TNF-α). A portion of the signaling pathway elicited by TNF-α is shown. Activation of the TNF-α receptor leads to activation and phosphorylation of I κB kinase (IKK). IKK in turn phosphorylates IκBα, which is then degraded via the proteosome. Free nuclear factor κB then translocates to the nucleus and activates gene expression. Given the role of TNF-α in triggering and maintaining states of insulin resistance, inhibition of its actions may prove to be an amendable therapy for type 2 diabetes. This can occur either at the level of circulating TNF-α, targeted with antibodies or soluble receptors, or at the level of its action on target tissues. Because the obligatory activation of NFκB signaling pathways is required for its biologic effect, inhibition of NFκB activity may be one way to counteract the actions of TNF-α. Interleukin 6 IL-6 is a 26-kd protein known formerly as B-cell stimulatory factor, interferon-β 2, 26-kd protein, or hepatocyte stimulating factor (126). Its multiple aliases reflect the pleiotropic nature of its physiologic functions. To date, IL-6 is known to participate in B-cell maturation and immunoglobulin production, acute-phase protein production in hepatocytes, neural cell survival and differentiation, activation of the hypothalamic–pituitary– adrenal axis, and expansion of hematopoietic progenitors (126,127,128,129). Expressed at very low levels in many cell types, it is rapidly induced upon injury, bacterial or viral infection, and stimulation by inflammatory cytokines (126,130,131,132). IL-6 is secreted from human adipocytes (133,134). Adipose tissue– derived IL-6 may constitute a large portion of circulating IL-6 (in low pg/mL quantity) under basal, noninflammatory conditions (133). Adipose-derived IL-6 probably does not play a major role in mounting an immune response against pathogens. As described below, its likely physiologic function is to promote energy utilization. IL-6 is structurally related to another adipokine described earlier, leptin. Like leptin, IL-6 is a member of the hematopoietic cytokine superfamily, characterized by a four-alpha-helix bundle core structure (135). One receptor for IL-6 (gp130) also belongs to the cytokine receptor superfamily that includes leptin receptor. Also like leptin, IL-6 activates Stat3 through Janus kinases (129). Their functions are nevertheless very different because the receptors are expressed in different cells. The cytoplasmic domain of a subunit of IL-6 receptor (IL-6Rα) is not required for its cellular effects (129). Rather IL-6Rα presents IL-6 to the ubiquitously expressed gp130, forming a signal-transducing complex (129). The membrane-anchored IL-6Rα in the signaling complex can be substituted with the soluble IL6 receptor containing just the extracellular domain (129). Because gp130 is a shared signaling molecule common to IL-6 and related cytokines (leukemia inhibitory factor, oncostatin M, and ciliary neurotrophic factor), they exhibit overlapping activities (129). P.969 Plasma IL-6 is elevated in obese individuals and decreased in those with high insulin sensitivity (133,136,137,138). However, whether this is due to decreased expression or secretion of IL-6 from adipocytes remains to be determined. With weight loss achieved by a very low-calorie diet in a cohort of obese women, plasma IL-6 levels were reduced along with production of IL-6 by adipose tissue (138). Little is known concerning the regulation of IL-6 expression in adipose tissue. Evidence suggests that it is regulated by other cytokines and the nervous system. TNF-α can potently increase expression of IL-6 in cultured adipocytes (139,140). In addition, β-adrenergic agonists can stimulate secretion of IL-6 from either isolated human adipocytes or differentiated 3T3-F442A–cultured adipocytes (141,142,143). Glucocorticoids are negative regulators of IL-6 production (134,140,143). Increased IL-6 levels in obese individuals may serve to limit further weight gain. IL-6 can reduce adipose tissue lipoprotein lipase activity and increase lipolysis (140,144,145,146). This may contribute to the hyperlipidemia component of the acute phase response following infection (145). Although not experimentally proven, reduced adipose tissue lipoprotein lipase activity will likely restrain fatty acid uptake by adipocytes. IL-6 is known to act in the CNS as an anorectic factor (147,148) and thus may control hyperphagia. Lastly, IL-6 is a powerful stimulator of the hypothalamic–pituitary–adrenal axis (147,149) and basal metabolic expenditure. Taken together, the metabolic effects of IL-6 are consistent with its potential role as an antiobesity cytokine. Indeed, mice deficient in IL-6 exhibit obesity characterized by increased adipose tissue mass (150). Resistin Resistin was discovered as an adipocyte-specific mRNA downregulated by rosiglitazone treatment (151) and upregulated during adipocyte differentiation (152). Also known as FIZZ3, it belongs to a family of secretory proteins named FIZZ (found in inflammatory zone) or RELM (resistin-like molecules) that share a carboxyl-terminal motif characterized by 10 precisely spaced cysteine residues (153,154,155). The first identified member of this family, FIZZ1/RELMα, was discovered as a gene markedly upregulated in lungs of mice with allergic pulmonary inflammation (153). Mature resistin secreted from differentiated 3T3-L1 adipocytes is a disulfide-linked homodimer of 94–amino acid polypeptides (156). Although there are 10 cysteine residues in the C-terminal half of resistin, one single cysteine near the N-terminus is responsible for interchain disulfide-linked dimer formation (156). The initial finding that resistin expression is regulated by the antidiabetic drug rosiglitazone suggests a role for it in the pathophysiology of type 2 diabetes. Several lines of evidence support this hypothesis. Recombinant resistin decreases insulin-stimulated glucose uptake in cultured 3T3-L1 adipocytes (151,157) and L6 myocytes (158). Administration of resistin to mice impaired glucose and insulin tolerance (151). The antagonistic effects of resistin on insulin action are not restricted to muscle and fat cells. Infusion of resistin into rats during a euglycemic clamp resulted in decreased whole-body glucose utilization due to increased hepatic glucose production at both low and high insulin levels (159). Resistin-rich conditioned media from transfected COS cells markedly inhibited differentiation of 3T3-L1 preadipocytes into mature adipocytes, possibly as a feedback mechanism to limit further development of obesity (45). All of these activities are consistent with resistin being a negative regulator of insulin action. Whether resistin mediates insulin resistance associated with obesity and improvement of insulin action following thiazolidinedione treatment remains a matter of debate. Although some studies have reported an increase in resistin mRNA or protein expression (151,160), others showed decreased levels in many different rodent models of obesity and diabetes (161,162,163,164). Although thiazolidinediones consistently decreased resistin expression in cultured adipocytes, it is not clear if this occurs in vivo in adipocytes (165,166). In addition, the findings related to resistin expression patterns and tissue distribution in mice may not be extended to humans (165,166,167,168). Many humans do not appear to express resistin in adipose tissue, and there is no correlation between expression levels and body mass index (167,168). Humans also only have two homologues as compared with three for rodents (154). In summary, whereas resistin’s biochemical and physiologic activities qualify it as a true adipokine, its role in the development of obesity and associated insulin resistance remains to be resolved. Angiotensin Adipose tissue contains a local renin–angiotensin system (RAS) (169). Expression of angiotensinogen, the precursor of angiotensin I, has been described in human and rodent adipose tissue as well as in cultured adipocyte cell lines (170,171,172,173,174). Although it is not clear if adipose tissue expresses renin, an enzyme capable of converting angiotensinogen into angiotensin I, its activity has been demonstrated in adipose tissue and cultured adipocytes (170,175). This could occur via uptake of circulating renin by adipose tissue. Angiotensin-converting enzyme, responsible for processing angiotensin I to angiotensin II, is also present in adipose tissue (170,171,172,175). The physiologic significance of the adipose RAS is not well understood. Expression and secretion of adipose angiotensinogen was reduced by fasting and increased with refeeding (176). However, it is not clear if expression of angiotensinogen or other components of the adipose RAS is elevated in obese humans (177). Thus, although obese individuals are prone to develop hypertension, it is not clear if this is due to dysregulation of adipose RAS. It is likely that adipose RAS regulates adipocyte differentiation and local blood flow. Adipocytes contain both AT1 and AT2 subtypes of the angiotensin II receptor (169). Angiotensin II may regulate adipocyte differentiation through stimulation of prostacyclin production from adipocytes (169,178). However, whether the effects of angiotensin II on adipose differentiation are stimulatory or inhibitory appear to depend on the particular model system used (169,178,179). Given the well-documented P.970 ability of angiotensin II to affect blood pressure, adipose RAS most likely contributes to regulation of local blood flow within adipose tissue. Through this mechanism adipose RAS may indirectly regulate rates of fatty acid release or reesterification and possibly influence circulating FFA levels, a key determinant of insulin sensitivity (176,180). Adipsin and Acylation- Stimulating Protein Adipose tissue can secrete the three early components of the antibody-independent alternate complement cascade: C3, factor B, and adipsin/factor D (181). During the activation of the alternative complement pathway, small amounts of C3b, a proteolytic fragment of C3, undergo conformational changes when bound to suitable cell surface receptors (182). This allows C3b to bind factor B and makes factor B susceptible to cleavage by factor D, also known as adipsin (182,183). The complex of C3b and a fragment of factor B, Bb, is the C3 convertase that cleaves C3 into C3a and C3b. C3b generated can interact with additional factor B to amplify this process or with C3 convertase to generate C5 convertase and membrane attack complex. C3a is an anaphylatoxin that stimulates histamine release (184). Although the proximal portion of the complement pathway is activated locally in adipose tissue in absence of pathogens, C5 and thus the late membrane attack portion of the cascade is absent (181). This suggests a non–immunity-related role for complement activation within adipose tissue. Adipsin was originally identified as an adipocyte-secreted protein upregulated during differentiation (185). Adipose tissue is a major production site for adipsin, although it is also expressed in other tissues (183,185). Expression of adipsin in adipose tissue and plasma adipsin levels were reduced in obese mice with disrupted hypothalamic leptin signaling (ob/ob, db/db, and newborn mice injected with monosodium glutamate), but not in obese rats fed a high-fat diet (186). In obese humans, serum and adipose-secreted adipsin levels were normal (187) or only mildly elevated (188). Serum levels of C3 and factor B were increased in obese individuals, but became normalized following weight reduction (187). Levels of adipsin, C3, and factor B were decreased in anorectic patients experiencing weight loss (187,188). These data suggest that adipose-derived C3, factor B, and adipsin have a role in regulation of energy balance in addition to an innate immune response. Altered adipsin expression in obesity and starvation indicates a possible function in lipid metabolism and energy homeostasis (185). Adipsin’s role in lipid metabolism became apparent when it was shown that the adipose-derived and anabolic acylation stimulating protein (ASP) is identical to the previously characterized C3a without the C-terminal arginine (C3adesArg) (189). ASP/C3adesArg is generated from C3a by carboxypeptidase B (190) and does not have the anaphylatoxic activity associated with C3a (184). ASP was purified from human plasma as a small basic protein with an actual molecular weight of 8,933 daltons determined by ion spray mass spectrometry (182,191). The concentration of ASP in mouse plasma is approximately 2 nM (190). Its major cellular action is to promote triglyceride synthesis by stimulating esterification of FFAs, as well as glucose uptake (189). This effect of ASP was seen in many types of cultured cells, but was most prominent in differentiated or primary adipocytes (182). Intraperitoneal injections of ASP accelerated removal of postprandial plasma triglycerides, FFAs, and glucose in normal, ob/ob, and db/db mice after fat loading (192,193). ASP is proposed to play a role in chylomicron metabolism because expression of C3, the precursor of ASP, is increased in cultured adipocytes in the presence of chylomicrons (194). Together these data support the hypothesis that ASP is an important regulator of the size of adipocyte triglyceride depots. Presumably ASP exerts its effects on triglyceride synthesis via an as yet unidentified plasma membrane surface receptor. Radiolabeled ASP demonstrates specific and high-affinity binding to adipocytes and fibroblasts (190). Stimulation of FFA esterification by ASP is associated with increased activity of diacylglycerol acyltransferase (195). Activities of other lipogenic enzymes were not affected (195). ASP can also stimulate lipogenesis by increasing glucose transport to supply the glycerol moiety of triglyceride (196,197). Phorbol 12myristate 13-acetate (PMA), a protein kinase C activator, can increase triglyceride synthesis in adipocytes similar to ASP (198). Additional support for a role of protein kinase C in the signaling pathway responsible for ASP-induced lipogenesis came from the observation that triglyceride accumulation by ASP can be blocked by inhibitors of protein kinase C (198). Despite the studies summarized above, whether dysregulation of ASP can contribute to development of obesity or type 2 diabetes remains unclear. Plasma ASP was found to be increased in obese humans and decreased with fasting (190). In addition, genetic ablation of the ASP precursor C3 in mice resulted in impaired triglyceride clearance and reduced adipose tissue mass (199). However, there is little difference in plasma ASP between db/db and control mice (190), suggesting ASP does not contribute to the obese and insulin-resistant phenotype of these mice. Transforming Growth Factor-β Transforming growth factor-β (TGF-β) is the prototypical member of a large secreted cytokine superfamily that includes bone morphogenic proteins, activins, anti-mullerian hormone, and myostatin. Three functionally nonredundant TGF-β isoforms are present in mammals: TGF-β1,2, and 3 (200,201). TGF-β is synthesized and secreted as an inactive complex that also contains latency-associated peptide, the propeptide generated by proteolytic cleavage of TGF-β precursor, as well as latent TGF-β binding proteins. Because both TGF-β and its receptors are ubiquitously expressed, an important activation step in initiating its responses is the release of the active form (202). Biologically active TGF-β can be released by either the matrix glycoprotein thrombospondin-1 or plasmin-mediated cleavage of the latent complex (200). The bioactive TGF-β is a 25-kd homodimer of two 12.5-kd monomers covalently linked by a disulfide bond (201). Actions of TGF-β are initiated by binding to type 2 TGF-β receptor (200). Subsequent recruitment of P.971 type 1 TGF-β receptor into the TGF-β/type 2 receptor complex then results in serine/threonine phosphorylation of the Smad2 or Smad3 transcription factors (202). TGF-β regulates a vast array of cellular processes that include differentiation, proliferation, and migration during embryonic development as well as wound repair and angiogenesis in adults (200,201,202). Due to its pleiotrophic effects, it is difficult to interpret the physiologic function of TGF-β in adipose tissue. Expression of TGF-β is elevated in adipose tissues of obese humans (203) and ob/ob and db/db mice (204). It is not clear what causes TGF-β to be increased in obese adipose tissue. To date, TNF-α is the only factor that has been shown to increase TGF-β production in adipocytes (204). Current data are consistent with elevated TGF-β as both a cause of obesity and a response to limit further obesity. TGF-β potently induces deposition of extracellular matrix by increasing the production of its constituent proteins and decreasing the production of degradation enzymes (200). Additionally, TGF-β increases the expression of PAI-1 (described below in detail), which is an inhibitor of many enzymes that degrade the extracellular matrix (4,200,204,205,206). TGF-β induction of extracellular matrix may serve to maintain the integrity of adipocytes as they grow ever larger (up to several times their original size) to accommodate increased triglyceride accumulation. TGF-β is also an established stimulator of angiogenesis (200). Appearance of newly formed adipocytes during expansion of adipose tissue mass also demands increased angiogenesis in addition to synthesis of extracellular matrix. Induction of extracellular matrix and angiogenesis by TGFβ suggest that its elevated expression contributes to growth of adipose tissue and development of obesity. In contrast to its positive effects on extracellular matrix deposition and angiogenesis, TGF-β may negatively control adipose development by inhibiting adipocyte proliferation and differentiation. The addition of small amounts of TGF-β (5 pM) to human or mouse preadipocytes in culture can completely block their differentiation into mature adipocytes (207,208). Unlike TNF-α, treating adipocytes already undergoing differentiation with TGF-β does not cause them to dedifferentiate (208). However, TGF-β can rapidly decrease expression of many adipose-specific genes including glycerol-3-phosphate dehydrogenase and PPAR-γ in fully differentiated adipocytes (208,209,210). These data suggest that elevated adipose TGF-β levels in the obese state serve to limit growth of mature adipocytes and differentiation of preadipocytes. Such inhibitory effects of TGF-β on adipocyte growth and differentiation appear to contradict the positive effects of TGF-β on angiogenesis and extracellular matrix synthesis. This is analogous to the dual roles of TGF-β as tumor suppressor and metastasis promoter (200). Clearly, further studies are required to resolve the role of TGF-β in the regulation of adipose tissue development and maintenance. Plasminogen Activator Inhibitor-1 Because cardiovascular dysfunction is closely associated with obesity, there may be adipokines that can regulate hemostasis. PAI-1 is a heavily glycosylated blood and extracellular matrix protein of approximately 50 kd belonging to the serine protease inhibitor (SERPIN) family (211). Although it is widely expressed, only liver, smooth muscle, platelets, and adipose tissue are thought to contribute to circulating PAI-1 (211). Although both cultured and primary adipocytes could produce PAI-1 (212,213), much of adipose-derived PAI-1 in plasma appears to be secreted from the stromal cells within the adipose tissue (213,214). More than 90% of PAI-1 in blood is stored in platelets (215), and the plasma concentration of PAI-1 is only about 400 pM (215). Its major function in blood is to inhibit removal of fibrin blood clots (fibrinolysis) by plasmin protease. PAI-1 performs this function by forming a stable complex with tissue-type plasminogen activator (t-PA) and prevents it from activating plasminogen into plasmin (214,216). PAI-1 also inhibits urokinase-type plasminogen activators (u-PA) near the plasma membrane of macrophages and endothelial cells (214,216). There it limits degradation of extracellular matrix and activation of metalloproteinases by plasmin (214,216). Independent of its plasmin inhibitor activity, PAI-1 can also regulate cell migration and adhesion via its interactions with vitronectin and the CD87 urokinase receptor (217). Unlike leptin or TNF-α, PAI-1 does not have a signal-transducing receptor. PAI-1 nevertheless is an important metabolic regulator secreted from adipose tissue. High plasma PAI-1 levels are correlated with increased incidence of cardiovascular disease (218). In conjunction with hyperinsulinemia and hypertriglyceridemia, PAI-1 is an important risk factor for coronary artery disease (215,218). Serum PAI-1 levels are elevated in obese humans and animals (203,218,219). Elevated PAI-1 levels may be a link responsible for the high incidence of cardiovascular disease in obese individuals (218,220). This is an attractive hypothesis that remains to be vigorously tested. First, although the contribution from adipose tissue to the circulating PAI-1 pool must be quantitatively defined, two observations support the idea that adipose-derived PAI-1 contributes significantly to this pool. Visceral, but not subcutaneous, adipose depots are associated with plasma PAI-1 levels (220,221,222). Coincidentally, PAI-1 expression is increased in visceral but not subcutaneous depots of obese humans and animals (222). However, whether visceral adipose tissue secretes more PAI-1 than do subcutaneous depots remains controversial (218). The etiology of elevated PAI-1 in obesity is poorly understood. Plasma PAI-1 levels can be upregulated by a variety of factors: insulin, angiotensin II, glucocorticoids, lipopolysaccharide, TNF-α, and TGF-β (211,215,218). Many of these factors are also upregulated in obesity. Consistent with its effects on other tissue types, TGFβ is a potent inducer of PAI-1 expression and secretion in adipose tissue (211,215,218). Whether TNF-α can increase adipose tissue PAI-1 is controversial (218). Effects of TNF-α on PAI-1 production may be mediated through increased TGF-β or IL-6 levels from adipocytes, possibly explaining the discrepancies among various reports. As described above, obesity is associated with elevated expression of both TNF-α and TGF-β in adipose tissue (4). These two molecules are likely to be responsible for increased adipose tissue PAI-1 levels in obese humans and animals via autocrine or paracrine interactions. Within adipose tissue, an increase in PAI-1 production may be necessary to ensure the integrity of P.972 extracellular matrix by inhibiting its degradation; this will protect the enlarged and more fragile adipocytes characteristic of the obese state. These data suggest that the increased circulating PAI-1 in obesity is a by-product of this proposed local function of PAI-1. If so, the cardiovascular component of the insulin resistance/metabolic syndrome may be seen as a consequence of the need to maintain the abnormally large adipose size in obesity. Tissue Factor In addition to PAI-1, another potential adipokine that can regulate hemostasis is tissue factor (TF). Also known as thromboplastin, TF is a type 1 transmembrane glycoprotein that initiates the extrinsic pathway of blood coagulation (223). Structurally it belongs to the class 2 cytokine receptor family that includes receptors for interferon-α and IL-10 (224). Even though TF is a transmembrane protein, it is found in circulating blood as embedded in small phospholipid-enveloped vesicles shed by cells (225). Expressed in vascular adventitia, connective tissue, skin epithelium, and many other tissue types, the expression pattern of TF has been described as an “envelope” that becomes activated upon rupture of the vasculature (223,226). TF is normally not exposed to blood. When it does, upon vascular injury, it binds to factor VII and facilitates conversion of the zymogen to the active serine protease VIIa (227). The TF-VIIa complex then forms a ternary complex with factor X where it is proteolytically cleaved by VIIa to become the active Xa (227). The active factor Xa then converts prothrombin to thrombin, which in turn cleaves fibrinogen to expose binding sites for other fibrinogen molecules and form a fibrin polymer. In addition to coagulation, TF is involved in the inflammatory response, angiogenesis, and extravasation of leukocytes and tumor cells (223). These effects are mediated through a family of seven-transmembrane domain and G protein–coupled protease-activated receptors (PARs) (228). These receptors become activated upon removal of an autoinhibitory Nterminal region by specific serine proteases. TF facilitates PAR activation directly by stimulating the proteolytic activity of the bound factor VIIa and also indirectly by generation of active factor Xa and thrombin proteases (227,228). TF is expressed in both adipocytes and stromal vascular cells within adipose tissue (229). TF mRNA levels in both cell types were markedly increased in ob/ob mice (229). Like PAI-1, the increased adipose TF in obese animals may reflect a need to better protect the vasculature of an adipose tissue containing the larger and more fragile adipocytes characteristic of obese animals (229). Through PAR, the elevated TF may be involved in the increased angiogenesis necessary for the enlargement of adipose tissue (229). The observation that TGF-β can increase TF expression in adipocytes is consistent with its role as stimulator of extracellular matrix and angiogenesis (229). In addition, TF has been shown to induce the expression of vascular endothelial growth factor (VEGF) in tumor cells (230,231,232). Taken together, these data indicate that TF is a true adipokine acting in a paracrine or autocrine fashion. In addition to adipose tissue, expression of TF is also elevated in brain, lung, kidney, heart, and liver of ob/ob as well as db/db mice (233). Such an increase may be due to increased TNF-α and TGF-β in the adipose tissue, both of which are known to activate TF expression (4,200,223) The physiologic significance of elevated TF expression in obese mice has not been firmly established. It is known that high factor VII activity is associated with increased cardiovascular risk, obesity, hypertriglyceridemia, and insulin resistance (218). Normally absent, inflammatory cytokines including TNF-α can induce the expression of TF in endothelial cells or monocytes (223). As described earlier, TF is found in circulating blood as embedded in small phospholipid-enveloped vesicles (225). The amount of TF in these circulating vesicles is elevated in patients with cardiovascular diseases and is thus thought to play a role in its development (225). Whether adipose tissue of obese individuals is one of the sources of these TF-rich vesicles is unknown. Increased TF on endothelial or monocyte surfaces as well as in microvesicles can promote deposition of arterial plaques. In this manner TF may serve as another potential molecular link, other than PAI-1, between coronary artery disease and obesity.