APL_Akaike_SI_revised

Supplemental Material
Fermi level pinning induced electrostatic fields and band bending at organic heterojunctions
K. Akaike, 1,a) N. Koch 1,2 and M. Oehzelt 1,2
1 Institut für Physik & IRIS Adlershof, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 6,
12489 Berlin, Germany
2 Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, Albert-Einstein-Str. 15, 12489
Berlin, Germany a) Electronic mail: kakaike@physik.hu-berlin.de
1
S1. UPS and XPS spectra of Au(111) substrate and MoO
3
film
Freshly prepared Au(111) substrate and 2 nm MoO
3
film were characterized by UPS and
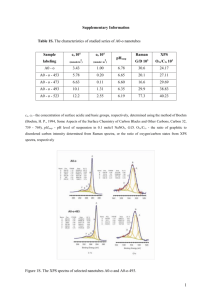
XPS. The cleanliness of the gold surface and a take-off angle located at normal emission were confirmed by surface states of Au(111) in the UPS spectrum (an asterisk in Fig. S1(a)). Upon the deposition of 2 nm MoO
3
, O 2p peak is observed at 3.8 eV (top spectrum in Fig. S1(a)).
The corresponding XPS spectrum in Mo 3d region shows a small fraction of Mo 5+ cations, which suggests the presence of a non-zero concentration of oxygen vacancies [Fig. S1(b)], while only one peak appears in O 1s region [Fig. S1(c)]. These observations are in good agreement with previous studies.
S1,S2
FIG. S1 (a) UPS spectra of a clean Au(111) substrate (bottom) and 2 nm-thick MoO
3
film (top).
An asterisk indicates the surface states of Au(111). (b) and (c) show Mo 3d and O 1s spectra of 2 nm MoO
3
film, respectively. The blue and red curves in (b) indicate the contributions from Mo
5+ and Mo
6+
cations, respectively.
2
S2. XPS spectra of ZnPc/C
60
interface formed on MoO
3
-covered gold substrate
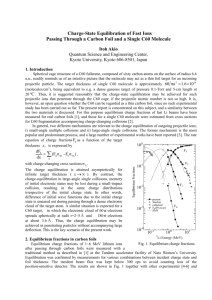
FIG. S2 Evolutions of XPS spectra of the ZnPc/C
60
interface in C 1s (a), Zn 2p
3/2
(b), and N 1s
(c) regions. Red and blue curves are assigned to carbon atoms of C
60
and ZnPc, respectively.
Ticks in (b) and (c) indicate positions of peak maxima. Dashed lines in (b) and (c) are guides to the eye to visualize the peak shifts.
S3. Calculation of percentage of charged molecules at the C
60
/ZnPc interface
In order to calculate the density of dipoles to explain the observed work function shift (
Wf) of 1.4 eV at C
60
/ZnPc interface [see Fig. 1(a)], the percentage of the charged molecules at the organic heterojunction was evaluated using the Helmholtz equation,
S3
∆WF = 𝑞 𝑝 𝑁
(S1) 𝜀
0 𝜀 with q being the elementary charge, N the interface dipole density, p the dipole moment perpendicular to the surface, ε
0
the vacuum permittivity, and ε the relative dielectric constant.
Assuming an localized dipole right at the interface we take the following values into account for the
Wf of 1.4 eV:
ε =
4, the distance of 1.1 nm between electron in the top C
60
layer and
3
hole in the interfacing ZnPc. The resulting charge density was found to be 4.5
10
–2
C m
–2
. The percentage of charged molecules is then calculated by dividing the charge density with the elementary charge, which result to 2.8
10 17 m
–2
. The percentage of charged molecules is then the ratio of the charge density and the density of molecules N . This results for ZnPc ( N= 2.0
10
18
m
–2
)
S4
to a fraction of 15% and for C
60
( N= 1.14
10
18
m
–2
)
S5
to 25%. Both fractions of charged molecules are large enough to be seen in UPS and XPS.
S3
On the other hand, if the counter charges are located at the gold electrode, the distance between the hole and electron increases to 14 nm (sum of thicknesses of MoO
3
and C
60
layers used in this study). The above calculation for ZnPc yields a fraction of only 1 % charged molecules. Such a small molecular fraction is not detectable with photoelectron spectroscopy.
Therefore, the latter assumption that electrons are transferred to the electrode is the only one compatible with our experimental results. Consequently, no localized interface dipole forms through the ZnPc pinning at the C
60
/ZnPc interface.
S4. Electrostatic potential calculations
As mentioned in the main text, the charge densities and electrostatic profiles were calculated to rationalize the experimental results. The density of states (DOS) of each molecular layer is modeled by two Gaussian functions with experimentally determined width, onsets of the HOMO and LUMO, and their respective orbital degeneracy. With the thickness of a layer of respective material and molecular number densities, the charge densities were calculated. The material parameters of ZnPc and C
60
are summarized in Table S1. The MoO
3
layer is treated as perfect insulator and therefore the DOS is set to zero. Only its dielectric constant of
r
of 14 is taken into account.
S6
Assuming the lateral homogeneity within respective layer, the calculations start by occupying all molecular layers in the organic heterointerfaces according to Fermi-Dirac statistics.
Then, the calculations proceed to numerically solve the 1D-Poisson equation on a grid of one molecular layer height, to calculate the layer-resolved electrostatic potential. Second iteration starts by occupying the DOS up to the Fermi level. The procedure cycles until a stationary solution for charge density and electrostatic potential is reached. A more detailed description of the model and its calculation are given in Ref. S7.
4
TABLE S1 Material parameters used in the electrostatic potential calculations
Ionization energy / eV
Band gap / eV
Electron affinity / eV
Degeneracy
ZnPc
4.8
a
1.9
b
2.9
c
HOMO: 1
LUMO: 1
C
60
6.4
a
2.4
4.0
d c
HOMO: 5
LUMO: 3
Full width at half maximum for HOMO and LUMO / eV
0.25 0.25
Dielectric constant 4.0
e
4.4
f
Molecular number density within one layer / m
–2
Height of one layer / nm
2.0 × 10
18 g
1.4
g a
Determined by UPS in this study. b Taken from Ref. S8. c Calculated by subtracting band gap from ionization energy. d Taken from Ref. S9. e Taken from Ref. S10. f
Taken from Ref. S11. g
Calculated from Ref. S4. h Taken from Ref. S5.
1.14 × 10
0.82
h
18 h
5
S4. Supporting reference
[S1] Irfan, M. Zhang, H. Ding, C. W. Tang, and Y. Gao. Org. Electron . 12 , 1588 (2011).
[S2] T. A. Papadopoulos, J. Meyer, H. Li, Z. Guan, A. Kahn, and J.-L. Brédas, Adv . Funct.
Mater. 23 , 6091 (2013).
[S3] P. Amsalem, J. Niederhausen, A. Wilke, G. Heimel, R. Schlesinger, S. Winkler, A. Vollmer
J. P. Rabe, and N. Koch, Phys. Rev. B , 87 , 035440 (2013).
[S4] JCPDS card no. 21-1986 and 39-1882.
[S5] D. L. Dorset and M. P. McCourt, Acta Cryst. A 50 , 344 (1994).
[S6] M. Sayer and A. Mansingh, Phys. Rev. B 6 , 4629-4643 (1972).
[S7] M. Oehzelt, N. Koch, and G. Heimel, Nat. Commun.
5 , 4174 (2014).
[S8] S. H. Park, J. G. Jeong, H.-J. Kim, S.-H. Park, M.-H. Cho, S. W. Cho, Y. Yi, M. Y. Heo, and H. Sohn, Appl. Phys. Lett. 96 , 013302 (2010).
[S9] Y. Tanaka, K. Kanai, Y. Ouchi, and K. Seki, Chem. Phys. Lett . 441 , 63 (2007).
[S10] I. Murtaza, I. Qazi, K. S. Karimov, and M. H. Sayyad, Physica B 406 , 533 (2011).
[S11] A. F. Hebard, R. C. Haddon, R. M. Fleming, and A. R. Kortan, Appl. Phys. Lett . 59 , 2109
(1991).
6