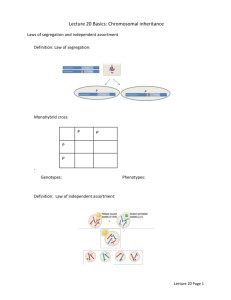
Practical_recombination
advertisement

DTC module in Bioinformatics: Nonparametric detection of recombination
Gil McVean and Simon Myers
In this practical we are going to look at one way of inferring the impact of recombination on
a set of DNA sequences. It makes use of a very simple idea, introduced in the lecture, that
there is a specific pattern in genetic variation that can most easily be explained by historical
recombination. The pattern in question relates to two linked SNPs. We are going to look at
how you can generalise this observation to a data set with an arbitrary number of SNPs. In
doing so you will develop a technique for solving special classes of problems called dynamic
programming.
Detecting recombination events in empirical data
If the mutation rate is very low, such that repeat or back mutation is very unlikely, every
time all four possible combinations of two alleles at two loci are observed we can be sure
that a recombination event must have occurred between them. In the example below, the
data on the left (A) show a detectable recombination and the data on the right (B) do not.
A)
B)
You are going to develop (and implement) an algorithm for calculating a lower bound for the
minimum number of recombination events that must have occurred in the history of our
sample of sequences - we will call this Rmin (Hudson and Kaplan 1985).
In the above examples, it is obvious that (A) requires at least one recombination and (B)
zero. In the examples below, how many do you need?
A)
B)
C)
To answer this you need to consider every pair of sites. For every pair of sites that show
evidence of recombination (i.e. for which you observe the four possible haplotypes) draw a
line underneath them. Once you have done this for every pair, find the minimum number of
positions at which you need to place a recombination event to explain the data.
For larger data sets we need to develop a systematic way of calculating Rmin. A good
starting point is to identify all pairs of SNPs for which a recombination event is detectable
between them using the four-gamete test. For example, below, the lines indicate the pairs
of ‘incompatible sites’ (those which have a detectable recombination event).
To find Rmin, we need to put the fewest possible recombination events down such that all
pairwise constraints are satisfied (a linear programming problem). What is the answer for
the above example? Can you see how to generalise the algorithm to an arbitrary number of
SNPs?
If not, consider the following two examples.
A)
B)
Note that (B) is identical to (A), except that it has an extra SNP at the right. In (A), we only
need a single recombination event, but when we add the extra SNP to make (B), we
generate an incompatibility between the 2nd and the 4th SNPs, which could not be the result
of the same recombination event that generated the incompatibility between the 1 st and 2nd
SNPs (note that the incompatibility between the 1st and the 4th SNPs could already be
explained). More generally, by thinking about adding in one SNP at a time, and looking to
see if it generates incompatibilities that are not currently explained, we can work
progressively along the sequence.
Explain why the formula
𝑅𝑚𝑖𝑛(𝑖) = 𝑚𝑎𝑥𝑗<𝑖 {𝑅𝑚𝑖𝑛(𝑗) + 𝐼𝑖𝑗 }
provides a solution to Rmin, where i and j are indices for the SNPs and Iij is an indicator
function that takes the value 0 if there is no detectable recombination between SNPs i and j
and 1 if there is).
Implement an algorithm for calculating Rmin from empirical data (alleles coded as 0s and 1s)
and apply it to the human data sets ceu7q31.txt and yri7q31.txt you can find at
http://www.stats.ox.ac.uk/~mcvean/DTC/BIOINF/Practicals
These are 707 SNPs genotyped on 60 individuals from a population of European ancestry
(CEPH families from Utah) and 60 individuals from the Yoruba in Nigeria. You will also find a
list of the relative positions of the SNPs in the file positions7q31.txt. Note that at most
Rmin can increase by one from the previous SNP. This observation can be used to increase
the efficiency of the algorithm.
Answer the following.
i) What is the minimum number of recombination events required for the two data
sets?
ii) Would you expect the two populations to show different amounts of historical
recombination?
iii) Plot how the minimum number of recombination events increases as you add more
SNPs. Does recombination appear to be clustered in particular regions or spread
evenly? Do you see any differences between the populations?