tpj12447-sup-0012-Legends
advertisement

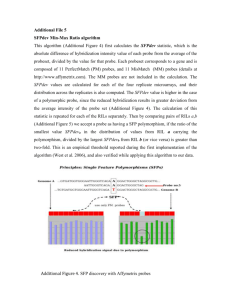
Supplementary Figure legends Method S1. Full experimental procedures. Includes methods for plant cultivation, transformation and analysis of physiology and morphology, BAC library, RIL and genetic map construction, CCP and BAC FISH, QTL, phylogeography, high throughput sequence analysis and NCBI Sequence Read Archive accession number SRR882054 for short read sequence data from this article. Fig. S1. Transmission distortion in the C. hirsuta Ox x Wa RIL population. The Chisquared statistic for observed allele frequency versus expected allele frequency is plotted against the genetic position on each linkage group. The positions of markers are shown by the blue dots. Significance thresholds for α=0.05 and α=0.01 are shown by the horizontal dashed and dotted lines respectively. Four relatively small regions of no more than two distorted linked markers were located on LG 1, 3, 7 and 8. The remainder mapped to 7 unlinked regions each including markers significantly distorted at p<0.01. The most severely distorted marker was located at 82.5 cM on LG 6, at the centre of a distorted region spanning 47 cM (p<0.05). At this locus, as well as at 3 other loci on LG 2, 5 and 8, Wa alleles were over-represented while at the remaining loci on LG 4, 6 and 7, Ox alleles were over-represented. To gauge whether these results may reflect inadvertent selection during construction of the RIL population, we studied segregation of alleles in the progeny of RIL46, which was heterozygous for markers 705_ and 475_3, in the distorted region on chromosome 4. We found only homozygous Ox alleles and heterozygotes in a 1:1 ratio for two markers in this region (N=146, Χ2 p<0.05), suggesting that genetic incompatibilities rather than inadvertent selection contributed to transmission distortion in this region. No significant linkage disequilibrium was found between unlinked distorted loci suggesting that higher order interactions or additive effects were responsible for the transmission distortion. Figure S2. Structure of C. hirsuta chromosomes CH6 and CH8 revealed by CCP using A. thaliana BAC contigs. A. thaliana BAC clones were arranged and labelled to distinguish seven genomic blocks (O, P, Q, R, V, W, and X) of the Ancestral Crucifer Karyotype. Chromosomes CH6 and CH8 were identified as a pachytene bivalent (left) and homologous chromosome pair (right). Arrowheads indicate centromeric regions. Scale bars:10µm. Figure S3. Proportion of coding region (CDS) length in A. thaliana genes covered by cDNA sequence of C. hirsuta homologues. 100% of CDS length is covered for 60% of C. hirsuta genes in the dataset. Proportion of A. thaliana CDS length is shown on the xaxis and frequency of C. hirsuta cDNA contigs is shown on the y-axis. Figure S4. Distribution of the number of gene ontology (GO) terms assigned to C. hirsuta cDNA contigs. Number of GO terms is shown on the x-axis and frequency of C. hirsuta cDNA contigs on the y-axis. Figure S5. Microsynteny between C. hirsuta, A. thaliana and A. lyrata. Colinearity along ~100-kbp of C. hirsuta, A. thaliana and A. lyrata genomic DNA was evaluated. Sequence of a single C. hirsuta BAC clone containing the CUP-SHAPED COTYLEDON2 (CUC2) gene located on C. hirsuta chromosome 8 was aligned to syntenic regions of A. thaliana chromosome 5 (AT9, chr5:21855k-21945k) and A. lyrata chromosome 8 (A. lyrata, chr8:16100k-16250k) using mauve genome alignment software (Darling et al. 2010). Six homologous blocks of sequence in the three genomes are colour coded and connected by lines. The height of bars within the sequence blocks reflects sequence identity. These six blocks of microsynteny were perfectly colinear in the two Arabidopsis genomes but the four smaller blocks were somewhat rearranged in C. hirsuta. Figure S6. Phylogeography of A. thaliana (Modified from (Beck et al. 2008). Statistical parsimony networks of A. thaliana nuclear PISTILLATA IVS 1 (a) and Atmyb2 (b) haplotypes. Each box represents a unique haplotype, with box size proportional to the haplotype frequency (small = 1–10 samples; medium = 11–49 samples; large = 50 or more samples). Connecting lines represent single mutations. Haplotypes described in (Beck et al. 2008) are filled in black. Haplotypes found both in this study and in (Beck et al. 2008) are outlined in red. New haplotypes found in this study are filled in red. Figure S7. Shoot branching scoring diagram. Branches that were found in the axils of rosette and cauline leaves, arising adjacent to the main stem, were scored as primary (1°) axillary branches, and branches found in leaf axils on these primary branches were scored as secondary (2°) axillary branches. Branches that arose adjacent to an axillary branch, in the axils of rosette and cauline leaves, were scored as accessory branches (shown in grey). Figure S8. Differential gene expression was determined from RNAseq reads of RIL founder accessions using fold change and read count to compute P-values. Significantly differentially expressed genes are shown in red. Figure S9. Distribution of C. hirsuta cDNA contig lengths. Contig length (bp) is shown on the x-axis and contig frequency is shown on the y-axis. Figure S10. Proportion of annotated cDNAs is lower for shorter contigs. C. hirsuta cDNA contig length is shown on the x-axis and the percentage of annotated sequences is shown on the y-axis. Figure S11. The distribution of average pairwise differences between coding regions in C. hirsuta and A. thaliana shown separately for the 1st, 2nd and 3rd codon positions. 3rd codon positions (green) are the most variable and even the slight difference in redundancy between the 1st (blue) and 2nd (red) codon positions is clearly visible in the distribution of divergence values, with 2nd codon positions being the least variable. Nucleotide divergence is shown on the x-axis and frequency on the y-axis. Table S1. Information for all molecular markers on the Ox x Wa F8 genetic map. Ox: number of RILs homozygous for Ox allele, Het: number of RILs heterozygous, Wa: number of RILs homozygous for Wa allele, !Ox: number of RILs not homozygous for Ox allele, !Wa: number of RILs not homozygous for Wa allele, Missing: number of RILs with missing genotype, Chi2: test statistic for observed versus expected allele frequencies in the RILs. Table S2. Accession list of the plant material included in the phylogeography study with detailed source information and ndhF-rpl32 haplotype numbers. Table S3. C. hirsuta genes that are differentially expressed between RIL founder accessions. SN = C. hirsuta stamen number QTL 1-8. Y = predicted stamen expression and N = not predicted stamen expression based on cited datasets. Table S4. Non-synonymous SNP variants in C. hirsuta RIL founder accessions, Ox and Wa. SN = C. hirsuta stamen number QTL 1-8. Y = predicted stamen expression based on cited datasets. Table S5. Differentially expressed genes between RIL founder accessions with predicted stamen expression in stamen number QTL regions. Y = predicted stamen expression based on cited datasets. Table S6. Non-synonymous SNP variants in C. hirsuta RIL founder accessions, Ox and Wa, found in stamen number QTL regions in predicted stamen-expressed genes. Y = predicted stamen expression based on cited datasets between RIL founder accessions. Table S7: C. hirsuta BAC probes used for FISH