density functional theory computations for design of salicylaldoxime
advertisement

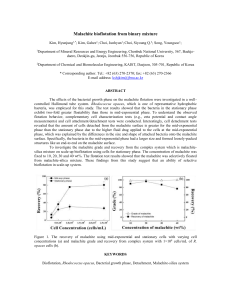
DENSITY FUNCTIONAL THEORY COMPUTATIONS FOR DESIGN OF SALICYLALDOXIME DERIVATIVES AS FLOTATION COLLECTORS FOR COMPLEX SULFIDE ORES Vinay Jain, Pradip and Beena Rai* Tata Research Development and Design Centre, Hadapsar Industrial Estate, Pune – 411 013, Maharashtra, India, *Corresponding author: Phone: +91 20 66086305. Email: beena.rai@tcs.com ABSTRACT The presence of oxide and oxidized minerals along with sulfides in mixed sulfide-oxide ores lead to losses in metal recovery during flotation with conventional thiol reagents. Chelating type reagents such as salicylaldoxime (SALO) and its derivatives are promising as flotation collectors for this system. We have earlier reported on the possibility of selective separation amongst copper, lead and zinc minerals using SALO derivatives with appropriate alkyl group substitution in the main chain (CM-SALO) or side chain (CS-SALO). The chain length of the substituent group as well as its position affects the selective interactions of SALO derivatives with metal ions both in the bulk as well as on the surface. We have performed density functional theory (DFT) computations to study interactions of SALO derivatives with Cu, Zn and Pb carbonate mineral surfaces, namely malachite [Cu2(OH)2CO3], smithsonite (ZnCO3) and cerussite (PbCO3). The objective of this study is to understand the underlying interaction mechanisms and use them in the design and development of better reagents for beneficiation of Cu-Pb-Zn ores. All computations were performed using the Quantum Espresso DFT package with plane wave ultrasoft pseudopotentials and Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation. The optimized bulk structures of the three minerals are in excellent agreement with experiments. The (010), (10-14) and (110) crystallographic cleavage planes were used for the interaction studies for malachite, smithsonite and cerussite respectively. The relative order of selectivity as per computed interaction energies was found to be: Malachite > Smithsonite > Cerussite. The main chain (CM-SALO) derivatives exhibit stronger interactions as compared to the side chain (CS-SALO) SALO derivatives. These findings are consistent with our experimental results reported earlier and provide crucial insight into the nature of the corresponding reagent-mineral surface interactions. Figure 1 – DFT-optimized geometry of CM-SALO – cerussite complex. Red, cyan, blue, grey, and yellow spheres represent O, H, N, Pb and C atoms respectively. KEYWORDS Salicylaldoxime, Density functional theory, Sulfide ores, Flotation, Malachite, Smithsonite, Cerussite