Tina – population admixture - University of South Carolina
advertisement

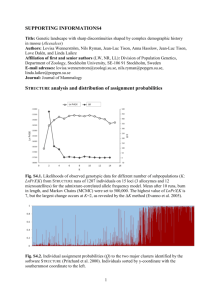
Drake 1 Kristina Drake Dr. Bert Ely BIOL 303, Section 501 5 November 2010 Population Genetics in Costa Rica Technological advancements in the last few centuries have increased dramatically the mobility of populations throughout the world. Thus, despite the physical barriers that separate the different continental populations, gene flow is common even over long distances. Immigration and interbreeding introduce new genetic variants into a population, and these additions to the gene pool create genetic admixture. One such example of an admixed population is Costa Rica1, a country with about four million people of diverse ancestry. Historical information about Costa Rica indicates that the nation’s population consists of genomic contributions from three continental populations, namely Africans, Europeans, and Native Americans1. This mixed pool of genetic information provides a unique opportunity to study the genes associated with vulnerability to common diseases, and as a foundation for this ultimate purpose, researchers have conducted analyses of the genetic composition and substructure of the Costa Rican population. In one study, scientists from the United States, France, and Costa Rica itself cooperated on a comprehensive population structure analysis of 1,301 women from the northern pacific region of Costa Rica (CR), known as Guanacaste1. They used admixture mapping to create an estimate of descent, by finding the degree of genetic admixture, measured by single nucleotide polymorphisms (SNPs). The women were genotyped on a custom Illumina InfiniumII iSelect panel containing 27, 904 tag SNPs. These tag SNPs are a subclass of SNPs that contain the Drake 2 majority of the information represented by the other SNPs in the surrounding region, based on the linkage disequilibrium (LD) structure2. Linkage disequilibrium is the degree of non-random association of alleles at multiple loci. These SNPs were selected according to a multiethnic tagging strategy which targeted candidate genes in European, African, and Asian HapMap reference populations. After standard quality-control filtering of the SNPs, 9,148 were identified as common among the CR samples and the four reference populations: the three HapMap samples plus Illumina 550 K Native American samples, all confirmed to be unrelated individuals1. Further filtering narrowed the results to 2,663 population structure inference SNPs with low background LD, which could otherwise interfere with the results. Five different programs were employed to analyze the structure and admixture of the CR population samples. The model-based STRUCTURE program was used to estimate the admixture proportions of the CR samples in comparison to the four continental reference samples. The CLUMPP program was used to compute the average admixture coefficients, and the DISTRUCT program plotted the range of resulting coefficients for all the samples. Principle components analysis was applied to determine a more in depth population structure, and the Haploview program performed haplotype analyses to estimate the average linkage disequilibrium block size for all the samples for comparison. The concepts behind these programs, primarily the STRUCTURE program, a modelbased method for inferring population structure from multilocus genotype data3. The number of ancestral populations in the data sample is assumed to be an unknown number K, and individuals in the sample are allocated to one of the K number of populations based on allele frequencies at different loci3. If the genotype of an individual appears to be admixed, it is assigned to multiple populations. Whichever value of K has the highest metric value of ∆K is the probabilistically Drake 3 optimal number of populations in the input data3. A subsequent study three years later modified this method to allow for the linkage between loci that develops in admixed populations, or the admixture linkage disequilibrium. This makes the program useful for admixture mapping, since it can detect more understated population subdivisions4. The improved algorithms were tested on admixed populations of African-Americans, and were used to study recombination in bacteria and genetic drift in fruit flies. This STRUCTURE program from these previous studies was applied to analyze the population structure in the CR sample. First, the program was run with various possible assumed K values to find the unknown value, and the optimum result was 4, meaning that there were four inferred ancestral populations for the pooled data of CR women and reference samples1. Since four distinct continental reference samples were entered into the program along with the CR samples, it was clear that the CR samples were not in themselves a separate ancestral population but rather an admixture. Then the STRUCTURE analysis was run again, using only that K value of 4, and the overall admixture proportions for the CR samples were calculated to be 42.5% European, 38.3% Native American, 15.2% African1. There appeared to be a 4% Asian influence, which was most likely the result of a few outlier individuals with strong Asian origins. These findings were consistent with the history of Costa Rican settlement, which was originally Native American with an influx of colonizing Europeans and West Africans1. To explore the population structure of Guanacaste in more detail, the researchers used a principal components analysis (PCA). PCA detects the primary axes of variation in the samples and projects them onto these axes graphically5. Three pairwise eigenvector comparisons were generated, and they supported the conclusion that the CR population is an admixture1. The CR samples spread between the reference population samples, primarily between the European and Drake 4 Native American data points, but skew noticeably towards the African samples as well. The second comparison clearly shows that the Asian population contributed very little to the CR genome1. The Asian data points lie in a clump entirely separate from the CR data points in the second graph of the eigenvector comparisons, and unlike the African population in the first comparison graph, there is little to no skewing in the direction of the Asian data points. The third comparison shows a separation in the North American samples between the Pima Indians and the more closely related Colombians and Mayans. The latter two Native American tribes more resemble the genetic composition of the CR samples. This association corresponds with historical knowledge about the Native American tribes; the Pima Indians typically resided much farther geographically from Costa Rican territory than the other tribes, and they did not migrate for colonization purposes into the area as did Europeans1. Since Asians and Pima Indians appeared to have contributed very little to the CR gene pool, the researchers refined their analysis by removing these samples from the input data. They also removed the few CR samples that contained Asian admixture greater than 10%, as these individuals were most likely outliers in the general composition of the CR population. The resulting PCA showed strong correlations between the EV projections and the admixture coefficients estimated by the STRUCTURE analysis. When the admixture coefficients for Europeans and Africans were compared in a histogram, a strong bimodality could be seen in the graphs. This double peak indicates that there are substructures in the CR samples1. The variability in the admixture coefficients, measured by the standard deviation, was 0.07 for Africans and 0.13 for Europeans. The low variability in the former suggests that the majority of the African admixing occurred during a relatively short period of time, early in the history of the Drake 5 Costa Ricans. The higher variability in the latter suggests that the European admixing occurred during a longer period of time, with a constant influx of European immigrants1. The African admixing fits with a relatively extreme model of admixture known as the Hybrid-Isolation model6, an idea proposed by Pfaff et al. in which the admixture takes place in a single generation. The European admixing fits with another extreme model from that study, known as the Continuous Gene Flow model6, in which the admixture takes place at a constant rate in every generation. These models were proposed as the result of research on linkage disequilibrium patterns in African American population samples from coastal South Carolina and Jackson, Mississippi, but can be applied to the Costa Rican population as well. In exploring possibilities for these substructures in Costa Rica, the researchers separated the samples into the different geographic regions of Guanacaste from which they originated. The three regions that seemed be correlated on the EV comparisons were the coastal cantons Santa Cruz and Nicoya and the inland canton Tilaran1. When compared to the projections of the reference populations, the Santa Cruz samples correlated towards a greater proportion of African admixture. Some of the Nicoya samples mixed in with those from Santa Cruz, but a large portion of them correlated towards a greater proportion of Native American admixture. The Tilaran samples correlated towards a greater proportion of European admixture. These findings are consistent with historical information that indicates that Nicoya was inhabited by several native tribes in the past, and that Tilaran had more recent European immigrants1. So, in conclusion, the substructures within the Guanacaste population appear to mirror the geographic location of the samples. Like the refined PCA, once the Asian and Pima samples were removed, the STRUCTURE analysis revealed more subtle nuances in the CR population. In the graph of the Drake 6 admixture proportions, there are two center points around which the CR samples are focused. The most samples concentrate around the point at 40% European, 40% Native American, and 20% African admixture. The second, smaller group of samples concentrates around the point at 60% European, 30% Native American, and 10% African admixture1. This suggests that the Guanacaste consists of two subpopulations with approximately these proportions of admixture, one with a high European percentage and the other with a lower but still predominant percentage1. Before studying these subpopulations further, the researchers investigated the potential effect of population stratification on false positive linkage signals in the admixture mapping. They sampled 200 subjects from each subpopulation and identified SNPs that have different genotype distributions between the two. The most differentiated locus, the rs1426654 SNP, was coincidentally in the gene neighborhood of an ancestry-informative marker associated with skin pigmentation, SLC24A51. It was chosen and used in a simulated scenario. In this hypothetical situation, the sample individuals with AA genotypes on that locus were chosen as cases and the other two genotypes were used as controls. An association test based on a logistic regression model with the additive genotype effect illustrated an inflation factor of 1.76 in the false positive rate, but with adjustments of the top two eigenvectors, the inflation factor dropped to 1.104. This simulated scenario represented an extreme case, but the strong population stratification effect could still be adequately corrected by the eigenvector adjustment1. The effects of population stratification on association studies and the importance of control selection were explored in a previous study7, and similar confounding effects were evident in the inflation factors. Data from the Cancer Genetic Markers of Susceptibility (CGEMS) project was used to determine the impact of population stratification on GWASs in Drake 7 breast cancer and prostate cancer in European Americans using various control selection strategies. These researchers determined that, despite the ancestral differences between controls and case subjects in a study with suboptimal control selection, the error can be reduced to an acceptable level with corrections for the inflation factor7. These are the corrections that were employed in the Costa Rican study to eliminate the population stratification error in admixture frequencies. The researchers then performed a Haplotype analysis to compare the average LD block size for each chromosome between the two subpopulations. Overall, the high-European admixture population had average block sizes that were 20% higher than the low-European population1. Because a greater proportion of admixture leads to a greater amount of LD, this data supported the hypothesis that the Guanacaste population as a whole was developed through a two-stage progression of admixture, in which the low-European group was formed first and then functioned as one of the ancestral populations for the high-European group to interbreed further with the European immigrants1. Then the LD magnitude and pattern in the CR samples was compared with those of the four reference population samples based on the original set of 9,148 SNPs. The TagZilla program was used to find the number of tagging SNPs required to cover the entire set of 9,148 SNPs with respect to different LD threshold values (r2=0.3, 0.5, 0.8). As the number of tag SNPs necessary decreases, the magnitude of the LD in that population increases1. Between the continental reference populations, the Africans required the greatest number of tag SNPs and the Native Americans required the least to encompass the same genomic area. In the CR population, less tag SNPs were necessary than in the Africans but more were needed than in the Native American, European, and Asian populations. Because of the multiethnic tagging strategy used in Drake 8 targeting these continental populations originally, the magnitude of LD detected for the reference samples may have been underestimated, however1. Overall, the results of this study on the population of Costa Rica indicate that the Guanacaste region is strongly admixed. The gene pool for this population is approximately 42.5% European, 38.3% Native American, and 15.2% African, with a nearly negligible 4% Asian influence. The variability in the admixture coefficients suggests that the integration of African genetic components into the Costa Rican population occurred early and for a brief period, whereas the integration of Europeans occurred steadily of a longer period of time. There are significantly different admixture coefficients between three geographic regions in Guanacaste, indicating a finer substructure to the population as a whole. The region can be split into two main subpopulations, one with a higher European admixture proportion (60%) and another with a lower proportion (40% European). The average linkage disequilibrium block size for the high-European subpopulation is 20% higher than that for the low-European subpopulation, suggesting that the low-European group was established first and then functioned as an additional ancestral population for the formation of the high-European group1. Works Cited [1] Wang, Z. et al. Genetic Admixture and Population Substructure in Guanacaste Costa Rica. PLoS One. 2010; 5(10): e13336. [2] "QuickSNP FAQs." Intute - Home. Johns Hopkins Medicine. Web. 04 Nov. 2010. <http://bioinformoodics.jhmi.edu/QuickSNP/FAQ.htm>. Drake 9 [3] Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. [4] Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. [5] McVean G. A Genealogical Interpretation of Principal Components Analysis. PLoS Genet. 2009;5(10):e1000686. [6] Pfaff CL, Parra EJ, Bonilla C, Hiester K, McKeigue PM, et al. Population structure in admixed populations: effect of admixture dynamics on the pattern of linkage disequilibrium. Am J Hum Genet. 2001;68:198–207. [7] Yu K, Wang Z, Li Q, Wacholder S, Hunter DJ, et al. Population substructure and control selection in genome-wide association studies. PLoS One. 2008;3(7):e2551.