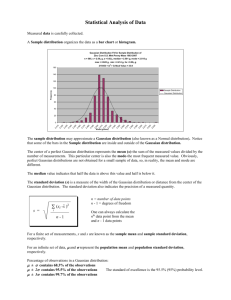
MFIELD command for SIGMA

$MFIELD command for SIGMA
****************************
! This Command is undergoing constant development, so contact me for the newest updates. The general method is described in Hu, H., Lu, Z., Yang, W.
QM/MM Minimum Free-Energy Path: Methodology and Application to
Triosephosphate Isomerase J. Chem. Theory Comput.
, DOI: http://dx.doi.org/10.1021/ct600240y
$MFIELD <newline>
SUBSystem atm1 atm2<newline>
JOBtype [FEN | FEP | NEB | MFE | QSM ] [<newline>
MDSTEP Num_of_MD_Step <newline>
FEPSTEP FEP_Step <newline>
CHARGESTEP Charge_Step <newline>
EQUIstep Equilibrium_Step <newline>
STEPSIZE Stepsize <newline>
SPRINGforce NEB_Spring_Force <newline>
DAMPfactor Damp_factor <newline>
MINIEND [SD | BFGS | DRMNG/PORT | GAUSS ] <newline>
QMTEMPLATE <newline>
QM_Template_File_Name <newline>
QMJOBNAME <newline>
QM_Job_File_Name <newline>
RECSTEP Charge_Record_Step <newline>
MICROOPT Micro_Traj_Step <newline>
MMTRAJFILE <newline>
MM_Trajectory_File_Name <newline>
MICROSTEP NEB_Spring_Force <newline>
INTERPolate conf_id <newline>
END <newline>
This command specifies "MEAN-FIELD" type of calculation in Sigma. Right now this command has to be used in combination with "$QM" command, since the purpose of this new simulation algorithm is to reduce QM calculation to minimal amount. Because the algorithm is designed for simulating a chain of
conformations, and thus naturally parallel, the Sigma program has to be compiled with MPI parallel mode, even though one may use single processor for optimizing single conformation for the reactant or product state.
SUBSystem is as the same as in " $QM " command. We need to specify the same region for QM calculation and MFIELD calculation at this time, and they have to be in the same order. (In the future, I may change this limitation, but
NOT NOW)
JOBtype specifies the type of calculation we want to perform.
" FEN " stands for " F reeE nergy-basedN EB" calculation.
" FEP " stands for " F reeE nergyP erturbation".
" NEB " stands for " N udgedE lasticB and".
" MFE " for " M inimalF reeE nergy(-Path)" (Compared with "Minimal Energy
Path" in Gaussian03).
" QSM " stands for " Q uadraticS tringM ethod" (reference: Burger, S. K.,
Yang, W., JCP, 124, 224102, 2006 ).
" FEN " is most often used.
MDSTEP says that how many steps of MD simulation are performed for the
"Mean-Field".
FEPSTEP says that we will do one step of FEP calculation for every
"FEP_Step" of MD simulation.
CHARGESTEP says that we will refresh charges for every "Charge_Step" of
MD simulation, for FEP calculation. The value of "Charge_Step" shall be the same as "QMFREQ" in " $QM " command.
EQUIstep says that for each "MD_Step" of MD simulation, the first
" Equilibrium_Step " steps will be omitted as equilibrium stage.
STEPSIZE specifies "timestep" for NEB calculation and minimization of endpoints. It shall be in the unit of "fs" (femto-second).
SPRINGforce specifies "spring force constant" for NEB calculation. It shall be in the unit of "kcal/mol/A^2" .
Obviously STEPSIZE and SPRINGforce are only meaningful for " NEB " type of calculations.
MINIEND decides which method shall be used to minimize the endpoint. " SD " stands for "Steepest-Descent", " BFGS " for "limited memory BFGS",
" DRMNG " for the "DRMNG" routine provide in the PORT library, and
" GAUSS " for Gaussian program. The use of "Gaussian" program is tricky and requires clear understanding of the method.
In short words, when this method is specified, the Sigma program will fork a
child process at the first step. The child process make a system call to Gaussian program, for which the input file shall specify the " External ", " Opt ", and
" NoSymm " keywords. This Gaussian program will make a "system" call again to an executable called "Gau_External" (you may have to write this code by yourself, or use the one I provided it in Sigma. You also need to make sure that this program is on your PATH.). The program "Gau_External" then communicates with the parent process of Sigma, to exchange coordinates and gradients. For this purpose, you will need another template file for creating
Gaussian input file, which is specified by " QMTEMPLATE ". If you have multiple Sigma jobs of the same type that run in the same directory, you may also want to specify different name for the "Gaussian" input, which is done by the keyword " QMJOBNAME ".
The idea of "free energy path" is that one can average out the effects of fluctuating MM conformations on the QM subsystem. The averaging process is achieved through MD simulation (of " MDSTEP "). Even though MD simulation is cheap, it is still unnecessary (and perhaps unwise) to carry out another round of MD simulation right after aone step of optimization on QM conformations.
Instead, we allow user to carry out multiple step of optimizations on QM subsystem within the same ensemble of MM conformations accumulated during the " MDSTEP " of MD simulations. This process is very much like the micro-optimization cycles performed in Gaussian program.
Controlling of this process is done by four keywords:
RECSTEP specifies how frequently we record MM charges for the use of computing QM charges (and response kernels if polarizable model is used).
This keyword relates to the "MMCHARGEFILE" keyword in " $QM " command.
MICROOPT specifies how frequently we record MM conformations for building a mean-field of MM potential. For every "Micro_Traj_Step", the program writes out the conformation into the file
" MM_Trajectory_File_Name" specified by " MMTRAJFILE ". Clearly this operation puts a huge burden on the IO of the computer system, so usually one should specify the file on a fast-accessing filesystem. Also Sigma program will NOT clean up those files after the run, the user has to figure out a way to remove those files. Otherwise, they will quickly fill up the disk.
MICROSTEP specifies how many "micro-optimization" step we will perform.
If one uses Gaussian program to optimize the end-point conformation, be sure that this number is larger that the optimization step limited in Gaussian input file.
!!! The meaning of the keywords " MICROOPT " and " MICROSTEP " is not so intuitive, be careful !!!
INTERPolate specifies which conformation ( j ) is going to be linearly interpolated from two neighboring conformations, j-1 and j+1 .