A HOMOLOGY BASED PCR CLONING STRATEGY TO ISOLATE A MONOTERPENE
SYNTHASE GENE INVOLVED IN -THUJAPLICIN BIOSYNTHESIS FROM CALLUS
CULTURES OF CUPRESSUS DUPREZIANA
A Thesis
Presented to the faculty of the Department of Chemistry
California State University, Sacramento
Submitted in partial satisfaction of
the requirements for the degree of
MASTER OF SCIENCE
in
Chemistry
(Biochemistry)
by
Soraya Ghasemiyeh
SUMMER
2012
©2012
Soraya Ghasemiyeh
ALL RIGHTS RESERVED
ii
A HOMOLOGY BASED PCR CLONING STRATEGY TO ISOLATE A MONOTERPENE
SYNTHASE GENE INVOLVED IN -THUJAPLICIN BIOSYNTHESIS FROM CALLUS
CULTURES OF CUPRESSUS DUPREZIANA
A Thesis
by
Soraya Ghasemiyeh
Approved by:
__________________________________, Committee Chair
Dr. Tom Savage
__________________________________, Second Reader
Dr. Mary McCarthy-Hintz
__________________________________, Third Reader
Dr. Nicholas Ewing
____________________________
Date
iii
Student: Soraya Ghasemiyeh
I certify that this student has met the requirements for format contained in the University
format manual, and that this thesis is suitable for shelving in the Library and credit is to be
awarded for the thesis.
__________________________, Graduate Coordinator
Dr. Susan Crawford
Department of Chemistry
iv
____________
Date
Abstract
of
A HOMOLOGY BASED PCR CLONING STRATEGY TO ISOLATE A MONOTERPENE
SYNTHASE GENE INVOLVED IN -THUJAPLICIN BIOSYNTHESIS FROM CALLUS
CULTURES OF CUPRESSUS DUPREZIANA
by
Soraya Ghasemiyeh
-thujaplicinalso referred to as hinokitiol, is a tropolone monoterpenoid with
antimicrobial and antifungal properties. It is responsible for the decay resistance of
heartwood of trees of the Cupressaceae. It is also used as an additive to toothpaste,
cosmetics, and foods in Japan and has scavenging activity against reactive oxygen
species and cytotoxic activity against several cancer cells lines. Here, Cupressus
dupreziana callus cultures were developed as an experimental system to isolate
thujaplicin biosynthetic genes. A homology-based PCR cloning strategy was
employed in an attempt to isolate a monoterpene synthase gene involved in thujaplicin biosynthesis.
_______________________, Committee Chair
Dr. Tom Savage
_______________________
Date
v
ACKNOWLEDGEMENTS
There are many people I would like to thank. First and foremost I would like
to thank Dr. Tom Savage, my research advisor, for all his help and guidance
throughout this process. He has been a great mentor and I am lucky to have had him
as my advisor. I would also like to thank my committee members, Dr. McCarthy and
Dr. Ewing, for their help and insightful comments.
Next I would like to thank John Disney and Ted Ferrera. Anytime I needed a
chemical or equipment John helped me find it and whenever an instrument wasn’t
working Ted was there to fix it. Chatting at the stockroom window was also
therapeutic. Thanks to all my fellow research students for the encouraging words and
friendship. A special thank you to Barbara Coulombe for helping me with my
presentation as well as keeping me sane towards the end of this process.
Finally I would like to thank my family. Thank you Hamid, for encouraging
and supporting me to do more - always. Thank you Naseem and Eemon, I was able to
focus on my studies because you are such great kids. I love you all more than I can
say. And to my parents who instilled the love of learning and always made me believe
I could do anything I set my mind to.
vi
TABLE OF CONTENTS
Acknowledgements ....................................................................................................... vi
List of Tables ................................................................................................................. ix
List of Figures................................................................................................................. x
Chapter
1. INTRODUCTION..………………………………………………………………….1
1.1 Decay Resistance of Wood From the Cupressaceae ............................................ 1
1.2 Tropolones ............................................................................................................ 3
1.3 Thujaplicins .......................................................................................................... 5
1.4 Terpene Biosynthesis............................................................................................ 7
1.5 Thujaplicin Biosynthesis .................................................................................... 11
2. METHODS AND MATERIALS ............................................................................. 19
2.1 Chemicals and Reagents ..................................................................................... 19
2.2 Initiation and Maintenance of Callus Cultures Providing
plicin ........... 20
2.3 Extraction and Derivitization Of-Thujaplicin ................................................. 21
2.4 GC-MS Analysis for -Thujaplicin and Other Monoterpenes ........................... 22
2.5 -thujaplicin Elicitation by Methyl Jasmonte .................................................... 23
2.6 RNA Isolation ..................................................................................................... 24
2.6.1 Equipment Preparation .............................................................................. 24
2.6.2 RNA Extraction and Analysis ................................................................... 24
2.7 Amplification of Monoterpene Synthase Gene Candidates ............................... 27
2.7.1 Degenerate Primers ................................................................................... 27
2.7.2 Synthesis of 5’ RACE-ready cDNA and 3’ RACE-ready cDNA ............. 28
2.7.3 Rapid Amplification of cDNA Ends (RACE) ........................................... 29
2.7.4 Gel Purification of PCR Products.............................................................. 30
vii
2.7.5 Cloning of Amplified Sequences ............................................................... 30
2.7.6 Sequencing and Analysis ........................................................................... 32
3. RESULTS AND DISCUSSION............................................................................... 33
3.1 Initiation and Maintenance of Callus Cultures ................................................... 33
3.2 Analysis of -thujaplicin and Other Monoterpene Content in Callus
Cultures............................................................................................................... 35
3.3 Elicitation of -thujaplicin by Methyl Jasmonate .............................................. 41
3.4 Isolation of RNA from C. dupreziana Callus Cultures ...................................... 42
3.5 Degenerate Primer Design .................................................................................. 48
3.6 Rapid Amplification of cDNA Ends (RACE) .................................................... 51
3.7 Conclusions ........................................................................................................ 68
References .................................................................................................................... 71
viii
LIST OF TABLES
Tables
1.
Page
Comparison of chemical components of Western Red Cedar,
Western Hemlock and Douglas Fir, adapted from (2). Numbers
expressed as a percentage of moisture-free weight of wood. ......................... 2
2.
Degenerate primers designed using CODEHOP. N=A+T+G+C,
D=G+A+T, H=A+T+C, R=A+G, W=A+T and Y=C+T. ............................. 28
3.
Codon degeneracy ......................................................................................... 48
4.
Degenerate primers for amplification of monoterpene symthase
cDNA. N=A+T+G+C, D=G+A+T, H=A+T+C, R=A+G, W=A+T
and Y=C+T. ................................................................................................... 51
5.
Gene specific primers..................................................................................... 56
6.
Most homologous sequence based on BLASTx and tBLASTx
searches of NCBI protein and nucleotide databases of 3’ RACE
products. ......................................................................................................... 61
7.
Most homologous sequence based on BLASTx and tBLASTx
searches of NCBI protein and nucleotide databases of degenerate
PCR products. ................................................................................................ 62
8.
Sequence analysis from gel purified 3’ RACE PCR...................................... 66
ix
LIST OF FIGURES
Figure
Page
1.
Structure of tropolone (2-hydroxy-2,4,6,-cycloheptatrien-1-one). ................. 4
2.
Some common tropolones and their structures from
Cupressaceae (20). ......................................................................................... 6
3.
Organization of terpene biosynthesis in plants. DMADP,
dimethylallyl diphosphate. IDP, isopentyl diphosphate. ............................... 9
4.
A mechanistic model of the monoterpene synthase reaction (12). ............... 10
5.
Label positions of -thujaplicin derived from [1-13C]-, [2-13C]-,
and [U-13C]-glucose with assumed labeled positions of GPP on
the way to -thujaplicin shown in parenthesis. Numberings of
geraniol carbons are shown with primes to distinguish them from thujaplicin carbons. Dots indicate 13C enriched, bold lines indicate
short-range coupling and double-sided arrows indicate long-range
coupling (31). ............................................................................................... 15
6.
Proposed pathway in -thujaplicin biosynthesis based on label
localization studies (31). ............................................................................... 15
7.
Proposed pathway in -thujaplicin biosynthesis (32). ................................. 16
8.
Proposed pathway for biosynthesis of -thujaplicin (17, 34)....................... 17
9.
Mechanism of first-strand SMARTer 5’ cDNA synthesis. .......................... 29
10.
Mechanism of first-strand SMARTer 3’ cDNA synthesis. .......................... 29
11.
Average callus size after six weeks. Values and error bars represent
means and standard errors of seven calli. ..................................................... 34
12.
C. dupreziana callus cutures......................................................................... 34
13.
Derivatization of -thujaplicin ..................................................................... 35
x
14.
Total ion chromatogram of derivatized 1.0 mg/mL -thujaplicin
with 1.0 mg/mL 1,3-napthalenediol as the internal standard. ...................... 36
15.
Mass spectrum ofderivatized -thujaplicin standard. .................................. 37
16.
Total ion chromatograms of derviatized -thujaplicin and 1,3
napthalenediol standards (top panel) and derivatized extract of
C. dupreziana callus (bottom panel). ........................................................... 38
17.
Mass spectrum of -thujaplicin standard after derivatization
(top panel) and derivatized -thujaplicin from extracts of
C. dupreziana (lower panel). ........................................................................ 39
18.
Total ion chromatogram of C. dupreziana callus culture
(1) -pinene, (2) 3-carene, (3) limonene, (4) terpinolene,
(5) carvacrol and (6) -thujaplicin. ………………………………………...41
19.
UV-VIS spectrum of isolated RNA from callus B. ...................................... 44
20.
Denaturing electrophoretic agarose (1.0 %) gel of RNA extracted
from calli. Lane 1: wheat grass RNA control, Lane 2 : callus A,
Lane 3 : callus B (From left to right). ........................................................... 45
21.
Total ion chromatogram of 1.0 mg/mL -thujaplicin with
0.1 mg/mL 2-napthol as the internal standard. ............................................. 46
22.
Mass spectrum of -thujaplicin standard. .................................................... 46
23.
Total ion chromatograms of -thujaplicin and 2-napthol standards
(upper panel) and C. dupreziana callus B extract with 0.025 mg/mL
2-napthol as internal standard (lower panel). ............................................... 47
24.
Sections of the aligned monoterpene synthase sequences with
RRX8W and DDXXD motifs highlighted and the primer blocks
marked above the sequences…………………………………………….....49
25.
Overview of 5' and 3' first-strand cDNA synthesis and 5' and
3' RACE PCR……………………………………………………………...53
xi
26.
Agarose gel electrophoresis of 3’ and 5’ RACE products.
Lane 1: 100 bp ladder (Invitrogen), Lane 2-4: 3’ RACE
with Block C, Lane 5-7: 5’ RACE with Block O primer,
Lanes 8-10: 5’ RACE with Block K primer, Lane 11:
100bp ladder (Invitrogen)…………………………………………………..54
27.
Agarose gel electrophoresis of 5’ RACE products.
Lane 1: Lamda III Hind digest (Invitrogen), Lane 2:
Callus B 5' RACE with Block O primer; boxed band
excised (~1500 base pairs), Lane 3: 100 base pair ladder
(Fisher Scientific), Lane 4: Callus B 5’ RACE with Block K
primer; boxed band excised (~850 base pairs). ............................................ 55
28.
Lane 1: 100 base pair ladder (Fisher Scientific), Lane 2:
Callus B 3’ RACE with GSP 1, Lane 3: Callus B 3’ RACE
with GSP 2 (excised bands boxed)………………………………………....57
29.
Lane 1: 100 base pair ladder (Fisher Scientific) Lane 2:
Fresh 3' RACE PCR product using GSP 2. .................................................. 58
30.
Colony screening via lysis and electrophoresis…………………………….59
31.
PCR screening of colonies with insert………………………...................... 60
32.
5' RACE PCR products. Lane 1: 100 bp ladder (Fisher
Scientific), Lane 2: 5’ RACE with Block K primer,
Lane 3: 5’ RACE with Block O primer………..…………………………..62
33.
3' and 5’ RACE PCR products. Lane 1: 100 bp ladder
(Fisher-Scientific), Lane 2: 3’ RACE with Block C primer,
Lane 3: 5’ RACE with Block K primer, Lane 4: 5’ RACE with Block O
primer. .......................................................................................................... 65
xii
1
Chapter 1
INTRODUCTION
-thujaplicin, also referred to as hinokitiol, is a tropolone monoterpenoid with
antimicrobial and antifungal properties. It is responsible for the decay resistance of
heartwood of trees of the Cupressaceae (1, 2). It is also used as an additive to
toothpaste, cosmetics, and foods in Japan (3, 4), has scavenging activity against
reactive oxygen species (5) and cytotoxic activity against several cancer cells lines (6).
Here, I discuss its role in the decay resistance of woods and how its antimicrobial
activity has led to interest in understanding its biosynthetic origin to enable its
production using biotechnology.
The goal of this study was to initiate and maintain callus cultures that actively
produce -thujaplicin in order to isolate the RNA. An RNA isolation protocol was
developed for callus cultures and once isolated, the RNA was used in first-strand
complementary DNA (cDNA) synthesis followed by 5’ and 3’ RACE so as to
elucidate the DNA sequence of a monoterpene synthase gene used in -thujaplicin
production.
1.1 Decay Resistance of Wood From the Cupressaceae
Conifer trees from the family Cupressaceae (cedars and cypresses) are found
globally, with the exception of Antarctica, and are well known for their resistance to
decay (7). They have naturally durable softwood and traditionally the heartwood is
2
used in applications where decay resistance is necessary such as utility poles, fence
posts, shingles and exterior construction. This quality spurred studies in the first half
of the twentieth century demonstrating that the heartwood is rarely attacked by decay
fungi (8-10). What differentiates the heartwood of the Cupressaceae from heartwood
from other coniferous species is the high levels of extractives. As shown in Table 1,
the wood from Western Red Cedar (a member of the Cupressaceae) has nearly twice
the extractives as wood from other coniferous species, whereas levels of structural
components cellulose and lignin are similar.
Table 1. Comparison of chemical components of Western Red Cedar, Western
Hemlock and Douglas Fir, adapted from (2). Numbers expressed as a percentage of
moisture-free weight of wood.
Species
Western
Red
Cedar
Western
Hemlock
Douglasfir
Family
Cellulose
Hemicelluloses
Lignin
Total
Extractives
Cupressaceae
47.5
13.2
29.3
10.2
Pinaceae
48.8
14.7
28.8
5.3
Pinaceae
53.8
13.3
26.7
5.9
Extractives, also known as secondary metabolites, are not an integral part of
the cellular structure of the tree but rather are compounds soluble in neutral organic
solvents or water (11). Secondary metabolites do not participate directly in growth or
development of plants but have important roles in protection, plant survival and plant
defense responses against insects, herbivores and microbial pathogens (12). They
3
include tannin, dyes, pitch, resins and gums that are responsible for the smell, taste
and color of the wood (2).
Extractives form at the heartwood/sapwood boundary using precursors that
have been transported from the phloem and sapwood. The concentration of
extractives varies greatly throughout the tree with an increase of extractives observed
with increasing distance from the pith (center) in heartwood and rapidly declining in
sapwood. Extractives are also found in higher concentrations toward the base of the
tree (2). Heartwood from younger trees contain a lesser amount of extractives than
heartwood from mature trees; however, the amount in younger trees is comparable to
the inner heartwood of mature trees, which is representative of the early growth
period. This suggests that the extractives are stable, and as the tree matures production
of extractives increases. This is consistent with the higher content of extractives found
in newly formed heartwood (2).
1.2 Tropolones
Some extractives found in the Cupressaceae family contain a tropolone
structure. Tropolones, are seven-membered aromatic compounds based on tropolone
(2-hydroxy-2, 4, 6-cycloheptatrien-1-one: Figure 1) that are relatively scarce in nature
(13, 14). Tropolones were not identified until 1945 when Dewar proposed this sevenmembered ring structure for stipitatic acid, a compound from Penicillium stipitatum
4
(15). This discovery gave birth to the field of non-benzenoid aromatic compounds
(14).
O
OH
Figure 1. Structure of tropolone (2-hydroxy-2,4,6,-cycloheptatrien-1-one).
Tropolones are of pharmacological interest due to their novel structures,
chemical properties and biological activities (13). These special characteristics can be
attributed to the 1,2 arrangement of the carbonyl and hydroxyl groups on the aromatic
seven-membered ring (15). It has been suggested that these biological effects may be
related to metal chelation between the carbonyl group at C-1 and the hydroxyl group
of C-2 (16).
Natural tropolones are found mainly in plants, specifically in Cupressaceae
and Liliaceae grass species, but a small number of bacterial and fungal tropolones
exist as well (13, 17). Many different tropolones are present in the heartwood of the
Cupressaceae family, some ubiquitous and others species dependent (Figure 2).
5
1.3 Thujaplicins
A group of tropolone compounds found in Cupressaceae family are known as
thujaplicins. The thujaplicins consist of three isomeric isopropyl tropolones known as
-thujaplicin-thujaplicin (hinokitiol) and -thujaplicin (Figure 2). These
thujaplicins are part of the volatile fraction of the heartwood extractives. Of the
thujaplicins, -thujaplicin is responsible for much of the decay resistance of the
heartwood. -thujaplicin inhibits blueing fungi more than the -thujaplicin and thujaplicin isomers or a synthetic fungicide, sodium pentacholorophenol, (8) and is
also a broad-spectrum antibacterial compound, inhibiting Gram-positive and Gramnegative bacteria (7). It has been shown that -thujaplicin can be used as a
postharvest treatment to prevent decay in some fruits and is a government-approved
food additive in Japan (4). -thujaplicin also activates the hypoxia-inducible factor,
HIF-1, pathway by inhibiting HIF-specific hydroxylases in human HepG2 hematoma
cells (ATCC HO-8065) and human HeLa cervical epithelium cells (ATCC CCL-2),
making it a potential treatment of ischemic diseases (18). Recent work on the topical
application of -thujaplicin with 5% zinc oxide on the skin of patients with atopic
dermatitis has shown inhibition, by interference of attachment, of Staphylococcus
aureus, including methicillin-resistant S. aureus which is of concern in hospital
settings (19). -thujaplicin has been shown to have cytotoxic activity against various
cancer cell lines including human stomach cancer KATO-III and Ehrlich’s ascites
carcinoma (6).
6
Figure 2. Some common tropolones and their structures from Cupressaceae (20).
7
The antimicrobial and antiproliferative properties of -thujaplicin have led to
an increased demand and interest in new methods of its production. One potential
source is to generate thujaplicins in fermentation organisms engineered with the ability
to synthesize these compounds. However, the biosynthetic pathway and the
corresponding genes that encode biosynthetic enzymes have yet to be elucidated.
Thus, there has been a recent focus on understanding the biochemistry and molecular
genetics of thujaplicin biosynthesis.
1.4 Terpene Biosynthesis
The volatile fractions of the extractives found in Cupressaceae are terpenes.
Terpenes are the most structurally varied class of plant natural products and are
derived by the fusion of isoprene units, branched 5 carbon units based on the
isopentane skeleton. Ten carbon (C10) monoterpenes contain two isoprene units,
fifteen carbon (C15) sesquiterpenes contain three isoprene units, and twenty carbon
(C20) diterpenes contain four isoprene units. Mono-, sesqui- and diterpenes are all
found in wood from the Cupressaceae (21).
Terpene biosynthesis begins with the formation of the precursors isopentyl
diphosphate (IDP) and its allylic isomer dimethylallyl diphosphate (DMADP). These
precursors are derived from one of two pathways. The mevalonate (MEV) pathway is
located in the cytosol and endoplasmic reticulum, and provides IDP and DMADP for
sesquiterpene biosynthesis, whereas the 2-C-methyl erythritol-4-phosphate (MEP)
8
pathway occurs in plastids and generates IDP and DMADP for monoterpene and
diterpene synthesis (Figure 3). These pathways differ in IDP precursors: acetyl-CoA
is the precursor to IDP in the MEV pathway, whereas pyruvate and D-glyceraldehyde3-phosphate are the precursors to IDP in the MEP pathway.
Of the terpenes, monoterpenes are best known as constituents of volatile
essences of flowers and the essential oils of herbs and spices. The biosynthesis of
monoterpenes (Figure 4) starts with geranyl diphosphate (GDP) being formed in the
plastid via the condensation of IDP and its isomer DMADP. Monoterpene synthases
subsequently catalyze the cyclization of GDP to monoterpene hydrocarbons, alcohols
or phosphates (Figure 4). Monoterpene synthases typically contain between 600 and
650 amino acid residues including an N-terminal transit peptide for plastid targeting
(22). Mechanistically, these enzymes ionize and isomerize GDP to form the tertiary
allylic isomer linalyl diphosphate (LDP). Subsequent ionization of enzyme-bound
LDP leads to cyclization to form a six membered ring (the -terpinyl carbocation)
which can then undergo additional reactions including rearrangements, oxidations,
addition of functional groups, or additional electrophilic cyclizations in the formation
of many monoterpene skeletons (12). The monoterpene synthase-catalyzed reaction
(Figure 4) is the first committed step in monoterpene biosynthesis and is responsible
for generating the skeletons that lead to all monoterpenes (23).
9
Figure 3. Organization of terpene biosynthesis in plants. DMADP, dimethylallyl
diphosphate. IDP, isopentyl diphosphate.
10
Figure 4. A mechanistic model of the monoterpene synthase reaction (12)
11
1.5 Thujaplicin Biosynthesis
-thujaplicin is a modified monoterpene with a tropolone structure (Figure 2)
found only in small amounts in the heartwood of mature trees. For this reason, it is
very difficult to acquire enough -thujaplicin for analysis or applications (20). For
example, each gram of sawdust from Thuja dolabrata only contains 200 g of thujaplicin (24). Thus, callus cultures have been developed to provide a more robust
and accessible experimental system to study tropolone accumulation. Calli are
proliferating masses of undifferentiated cells that have the potential to produce the
range of chemicals found in the parent plant (25). Callus cultures of Cupressus
lusitanica and Thuja occidentalis have been established and studied for many years
(26), and these calli have been shown to produce -thujaplicin.
De novo biosynthesis of -thujaplicin and other monoterpenes in calli can be
stimulated by treatment with methyl jasmonate or fungal extracts (26-28). thujaplicin accumulates initially upon elicitation in calli, and other monoterpenes, such
as 4-terpineol and 1,6-epoxy-4(8)-en-p-menthene-2-ol, are produced later. A feedback
regulation of -thujaplicin exists in elicited culture, with its methyl ether being formed
once a threshold of 40mg/L is reached suggesting that high levels -thujaplicin may be
toxic to callus cells (29).
12
Initial work to elucidate the biosynthetic pathway of -thujaplicin occurred
before it was recognized that -thujaplicin is a monoterpene. Because polyketidebased pathways that involve polymerization of acetate or malonate are also
responsible for a wide variety of natural product structures, early experiments were
designed to differentiate between potential polyketide and isoprenoid origins of thujaplicin.
A precursor feeding study with calli by Yamaguchi et al. (30) incubated yeastelicited C. lusitanica cells with [U-14C]glucose, [2-14C]mevalonate and [2C]malonate as substrates (30). Significantly more incorporation of label into -
14
thujaplicin was observed from glucose than mevalonate, implying formation through a
polyketide pathway. However, incorporation of radiolabel from [2-14C] malonate (a
more direct polyketide precursor) was not observed, refuting the polyketide pathway.
It should be noted that the early experiments were performed before it was recognized
that monoterpenes are formed from IDP generated by the MEP pathway rather than
from mevalonate. Thus, the incorporation of label from glucose (a precursor for the
MEP pathway) rather than from malonate or mevalonate is consistent with thujaplicins
being synthesized via a MEP-based isoprenoid pathway.
After the existence of the MEV pathway became well-established, an
experiment was developed that involved feeding radiolabeled [2-14C] mevalonate, [10C] geraniol and [U-14C] glucose to calli to confirm the isoprenoid origin of -
14
13
thujaplicin and establish whether the isoprene substrate arises from the MEV or MEP
pathway (31). Radiolabel from [10-14C] geraniol (which can be phosphorylated in
vivo to GDP) was incorporated into -thujaplicin more than 2 times greater than
radiolabel from [U-14C] glucose and more than 200 times greater than radiolabel from
[2-14C] mevalonate, indicating that the isoprenoid GDP is a precursor to -thujaplicin
(31).
Labeling experiments were then used to distinguish whether GDP incorporated
into -thujaplicin arises either from the MEV or MEP pathway. Isoprenoids produced
from labeled [U-13C] glucose through the MEP pathway contain three carbons from
the labeled glucose molecule incorporated into the final product that has a
characteristic coupling spectra observable in 2D INADEQUATE NMR. GDP was
shown to be a product of the MEP pathway due to the coupling patterns as well as
negligible incorporation of [2-14C] mevalonate into -thujaplicin.
Additional label localization studies using [1-13C] glucose, [2-13C] glucose and
[U-13C] glucose as precursors led to the development of a specific biosynthetic
hypothesis (Figures 5 and 6). Geraniol derived from [1-13C]- and [2-13C]- glucose via
the MEP pathway should be labeled at positions 1’, 5’, 9’ and 10’ and 2’, 3’, 6’ and 7’,
respectively (Figure 5). -thujaplicin derived from [1-13C]- and [2-13C]- glucose
showed increased 13C NMR measurements, indicating labeled carbons, at positions 3,
5, 7 and 9 and at 1, 4, 6 and 8, respectively (Figure 5). This data indicates that
14
rearrangement of the isopropyl group did not occur during -thujaplicin biosynthesis
because the C-4 and C-8 were enriched with [2-13C]-glucose feeding and the longrange coupling between C-4 and C-9 (or 10) was present in [U-13C]-glucose feeding
experiment. Therefore, C-3, C-4, C-8 C-9 and C-10 of -thujaplicin corresponds to C5’, C-6’, C-7’, C-8’ and C-9’ of geraniol, respectively. The C-2 of -thujaplicin is not
labeled by [1-13C]- or [2-13C]-glucose feeding, but does exhibit long-range coupling
with C-5 in the [U-13C]-glucose feeding experiment and must therefore be C-4’ from
geraniol. An adjacent pair, C-2’ and C-3’ (enriched by [2-13C]-glucose) in geraniol
was separated by an enriched carbon from [1-13C]-glucose feeding and the adjacent C3’ and C-10’ pair that have no long-range coupling with any other carbon in the [UC]-glucose feeding remain adjacent as C-1 and C-7 of -thujaplicin. Thus, the
13
methyl group at the 10’-position in geraniol splits the C-2’ and C-3’ bond and
therefore, C-1’, C-2’, C-3’ and C-10’ of geraniol correspond to C-5, C-6, C-1 and C-7
of -thujaplicin. From the findings a carbon skeletal rearrangement from geraniol to
-thujaplicin was suggested (Figure 6).
15
Figure 5. Label positions of -thujaplicin derived from [1-13C]-, [2-13C]-, and [U-13C]glucose with assumed labeled positions of GPP on the way to -thujaplicin shown in
parenthesis. Numberings of geraniol carbons are shown with primes to distinguish
them from -thujaplicin carbons. Dots indicate 13C enriched, bold lines indicate shortrange coupling and double-sided arrows indicate long-range coupling (31).
Figure 6. Proposed pathway in -thujaplicin biosynthesis based on label localization
studies (31).
16
Another study suggested 2-carene or terpinyl acetate as possible intermediates
in -thujaplicin biosynthesis (Figure 7). This study involved feeding cultures with six
unlabeled monoterpenes at different concentrations to investigate the relationship
between the monoterpenes and -thujaplicin biosynthesis. Addition of 2-carene or
terpinyl acetate promoted -thujaplicin accumulation by 2- 2.5 fold with respect to the
control whereas the other monoterpenes were inhibitory. These results led the authors
to propose two possible pathways, shown in Figure 7.
Figure 7. Proposed pathway in -thujaplicin biosynthesis (32).
17
Finally, another pathway for -thujaplicin biosynthesis has been proposed
based on the timing of accumulation of related monoterpenes (Figure 8). By
modifying the growth medium to prevent -thujaplicin biosynthesis, 1,6-epoxy-4(8)p-menthen-2-ol (33) rapidly accumulated whereas when the medium was modified to
increase -thujaplicin biosynthesis 1,6-epoxy-4(8)-p-menthen-2-ol remained at a basal
level suggesting that this novel monoterpene may be an intermediate in -thujaplicin
biosynthesis (34).
Figure 8. Proposed pathway for biosynthesis of -thujaplicin (17, 34).
Thus, to date, alternate proposals for the biosynthetic pathway to -thujaplicin
have been developed based on different types of experimental evidence. However,
these pathways are not definitive and no progress has been made towards isolating
genes encoding thujaplicin biosynthetic enzymes. Nonetheless, each proposed
18
pathway incorporates a monoterpene synthase-catalyzed reaction to produce the basic
monoterpene skeleton, with subsequent modifications to generate thujaplicin.
Other studies on monoterpene biosynthesis have employed cloning techniques
to isolate monotepene synthase genes. By isolating the monoterpene synthase genes,
not only is it possible to arrive at more conclusive pathways in monoterpene
biosynthesis but also enables transgenic manipulation to increase production of
essential oils or phytopharmaceuticals (12, 21, 35).
Here we employ a molecular genetic approach to isolate a monoterpene
synthase sequence involved in -thujaplicin production. The first goal was to
establish a tissue culture system that was actively producing -thujaplicin to be used
as a source of mRNA. The mRNA was isolated from the calli and consensus
monoterpene synthase amino acid sequences were used to design degenerate primers
for use in RT-PCR in order to amplify and isolate cDNA sequences encoding
candidate monoterpene synthase genes.
Once a monoterpene synthase-like sequence is isolated we can then use
portions of it as gene specific primers to get a full-length monoterpene synthase
cDNA. This can then be cloned in to a microbial vector to express the enzyme in
order to biochemically characterize and obtain correct functional identification of the
gene product or products. This will enable a better understanding of molecular
processes and characterization and may also enable microbial production of thujaplicin.
19
Chapter 2
METHODS AND MATERIALS
2.1 Chemicals and Reagents
Phytagel (plant cell culture powder), sucrose, Murashige and Skoog Basal Salt
mixture (MS), Gamborg’s Vitamin Solution 1000X, α-napthalene acetic acid (NAA),
6-Benzylaminopurine (BAP), bromphenol blue, polyvinylpyrrolidone (PVP), ethidium
bromide, -mercaptoethanol (BME), and 3-(N-morpholino)propanesulfonic acid
(MOPS) were purchased from Sigma-Aldrich (St. Louis, MO). Molecular biology
grade ethylenediaminetetraacetic acid (EDTA), Tris-HCl, aurintricarboxylic acid,
sodium acetate and lithium chloride were purchased from Calbiochem (Darmstadt,
Germany). Optical grade cesium chloride was purchased from IBI Scientific (Pesota,
IA). N,O-bis(trimethylsilyl)trifluoroacetamide with trimethylchlorosilane (BSTFA +
1%TMCS) was purchased from Thermo Scientific (Rockford, IL). 1,3-napthalenediol
was supplied from Matheson, Coleman and Bell (Norwood, OH); dithiothreitol (DTT)
from ACROS Organics (Geel, Belgium); and agarose, TBE buffer and formamide
from Fisher Biotech (Fair Lawn, NJ). Amberlite MB3 resin was purchased from
Mallinckrodt (St.Louis, MO) and formaldehyde from Spectrum (Gardena, CA).
SMARTer™ RACE cDNA Amplification Kit was purchased from Clontech (Mountain
View, CA), the pCR®8/GW/TOPO® TA Cloning® Kit and custom primers were
purchased from Invitrogen (Grand Island, NY), the 5 PRIME MasterMix from 5
20
PRIME (Gaithersburg, MD) and the Quantum Prep® Plasmid Miniprep Kit were
purchased from BioRad (Hercules, CA). Thiabendazol, diethylpyrocarbonate (DEPC)
and -pinene were purchased from MP Biomedicals Inc. (Solon, OH). All water used
in RNA isolation was treated with 0.1% v/v DEPC left overnight and then autoclaved
before use.
2.2 Initiation and Maintenance of Callus Cultures Providing thujaplicin
To screen a variety Cupressus species for the ability to produce callus cultures
capable of producing -thujaplicin, young branches of C. dupreziana, C. lawsonia, C.
macnabiana, C. nootkatensis and Thuja orientalis were obtained from the CSUS
arboretum and samples of C. lusitanica were obtained from the UC Davis arboretum.
Specimens from the CSUS arboretum were identified by botany professor Michael
Baad and the sample from the UC Davis arboretum was identified by Arboretum
Superintendant Emeritus Warren Roberts. Branch samples were cut from the trees and
placed on ice. Each sample was cut into 10 - 15 cm long pieces and surface-sterilized
by soaking in 85% (v/v) ethanol solution for ten minutes, followed by soaking in 20%
(v/v) bleach and SDS solution for thirty minutes. The samples were then soaked in
70% ethanol solution for ten minutes and subsequently rinsed three times with
sterilized deionized water. Each piece was then sliced into 1-2 mm thick slivers and
placed on 0.15% (w/v) phytagel agar plates containing 0.43% (w/v) Murashige and
Skoog salts (36), 10-5M BAP, 10-2M NAA, 0.1% (w/v) 1000X Gamborg’s Vitamin
21
Solution, 2.0% sucrose and 2.5 x 10-4 M thiabendazole at pH 5.5. These plates were
then sealed with parafilm and placed in the dark for two weeks. Once established, the
calli were then transferred onto MS plates excluding thiabendazole to encourage more
vigorous growth, and were replated every four to six weeks.
2.3 Extraction and Derivitization Of -Thujaplicin
To analyze -thujaplicin production in the established cultures, 0.5 g fresh
callus was pulverized in a mortar and pestle and one mL of ethyl acetate containing
1.56 x 10-4 M 1,3-napthalenediol as an internal standard was added. The resulting
paste was then placed in an airtight vial and the mortar and pestle was rinsed with
ethyl acetate and added to the paste and left overnight. The extract was subsequently
filtered through glass wool and the samples were dried on a vacuum concentrator on
medium heat. The dried samples had L of methylene chloride added to insure
complete removal of water and dried again. Finally, 150 L of BFSTA–1% TMCS
was added to the dried samples to generate the trimethylsilyl derivative. A sample of
6.09 x 10-4 M -thujaplicin with 1.56 x 10-4 M 1,3-napthalenediol was subjected to the
same procedure as a positive control. These solutions were placed in airtight vials until
analysis by gas chromatography-mass spectrometry (GC-MS).
22
2.4 GC-MS Analysis for -Thujaplicin and Other Monoterpenes
Gas chromatography-mass spectroscopy (GC-MS) analysis of derivatized thujaplicin standard and callus extracts were performed in electron impact mode with
an Agilent 7890 GC equipped with an Agilent 5975 MS. Separation was carried out
using a HP-5MS, (5%-Phenyl)-methylpolysiloxane, column, 0.25 mm diameter and
0.25m film thickness. Split injections of 1 L were used. The temperature program
was: 40 oC for 1 minute, followed by a temperature ramp of 20 oC/min to 150 oC, 30
o
C/min to 280 oC, finally holding at 280 oC for 7 minutes. The carrier gas was helium
at a linear velocity of 1.2 mL/min, and the injector was maintained at 250 oC .
Analyses were performed in full scan mode over the range of m/z 40-550. The MS
source and quadrapole were maintained at 230oC and 150oC, respectively.
Quantification of -thujaplicin was performed by GC-MS with the addition of
1.56x10-4 M 1,3-napthalenediol as the internal standard. The areas of total ion peaks
were integrated and levels of -thujaplicin present in different calli were calculated
based on comparison of peak areas to the known concentration of 1,3-napthalenediol.
To analyze other monoterpenes, calli were pulverized in a mortar and pestle
then steam-distilled with deionized water. The distillate was extracted with ice-cold
pentane, which was concentrated by evaporation with nitrogen gas and examined by
GC-MS using the same instrument and column as before except the temperature
program was 40oC for one minute, followed by a temperature ramp of 5 oC/minute to
23
200 oC, holding for 5 minutes. The compounds were identified by comparison to the
NIST database.
For direct analysis of -thujaplicin without derivatization, 2-napthol was used
as an internal standard. The calli were pulverized in a mortar and pestle, and 1mL of
ethyl acetate containing 1.73 x 10-4 M 2-napthol as an internal standard was added.
The resulting paste was placed in an airtight vial, and the mortar and pestle were
rinsed with ethyl acetate, which was added to the paste, which was then left overnight
at room temperature. The extracts were filtered through glass wool, and the samples
were dried on a vacuum concentrator on medium heat. The dried samples were mixed
with L of methylene chloride to insure complete removal of water and then dried
again. A sample of 6.09 x 10-4 M -thujaplicin with 6.94 M x 10-4 M 2-napthol was
subjected to the same procedure as a positive control. These solutions were placed in
airtight vials until analysis by GC-MS.
2.5 -thujaplicin Elicitation by Methyl Jasmonte
A 200
M solution of methyl jasmonate was made by adding 223.6 L of
methyl jasmonate to methanol for a final volume of 5 mL. Five L of the 200 mM
methyl jasmonate solution was then added to 5 mL of nanopure water for a final
200M solution and filter-sterilized through a 0.2m filter. A control solution was
made by adding 5 L of methanol to nanopure water and filter-sterilizing. Four C.
dupreziana calli were halved and transferred to fresh MS plates; one half was treated
24
with 1 mL of the control solution while the other half was treated with 1 mL of the
200 M methyl jasmonate solution. Each set of calli was incubated for a week at
room temperature before extraction, derivatization and GC-MS analysis.
2.6 RNA Isolation
2.6.1 Equipment Preparation
All glassware and nonplastic equipment was washed and rinsed three times
with nanopure water and oven dried at 185 oC overnight. All reusable plastic
materials, such as polypropylene centrifuge tubes, were washed and rinsed with
nanopure water followed by soaking in 3% H2O2 for 10 minutes and finally rinsed in
DEPC (0.1% v/v) treated water. Disposable plastics such as pipet tips and eppendorf
tubes were DNase and RNase free. All surfaces were wiped down with ethanol and
aseptic techniques employed.
2.6.2 RNA Extraction and Analysis
Six different protocols were used in an attempt to isolate RNA of sufficient
yield and purity for cDNA synthesis. Methods tried involved the Spectrum™ Plant
Total RNA Kit (Sigma-Aldrich) according to manufacturer’s protocol, the Spectrum™
Plant Total RNA Kit (Sigma-Aldrich) modified by adding 1% w/v PVP to lysis
solution as well as performing a 10% v/v cold ethanol precipitation step, a LiCl/hot
phenol extraction (37), isolation using TRIzol® (38), Direct-zol™ RNA Miniprep Kit
25
(Zymo Research) and a modified version of a protocol by Lewinsohn et al (39). Only
the Lewinsohn protocol afforded viable RNA from the callus cultures, however, high
quality RNA from fresh wheat grass was isolated with the Spectrum™ Plant Total
RNA Kit (Sigma-Aldrich), which was used as a control for electrophoretic analysis of
RNA .
Using the Lewinsohn protocol, C. dupreziana callus cultures containing thujaplicin, based on GC-MS analysis, were frozen in liquid nitrogen and ground to a
fine powder with a mortar and pestle. The powder was then added to the extraction
buffer (200 mM Tris-HCl, 300 mM LiCl and 10 mM Na acetate in DEPC treated
water, pH 8.5, containing 1% w/v PVP, 1mM aurintricarboxylic acid and 10 mM
ditihiothreitol) in polypropylene tubes and vortexed, followed by centrifugation at
5000 x g for 20 minutes at 4 oC. The supernatant was decanted and 1/30 volume (110
mM final concentration) of 3.3M sodium acetate buffer, pH 6.1, and 10% (v/v) of cold
ethanol were added. This was incubated on ice for 10 minutes to allow for
polysaccharide precipitation and then centrifuged at 5000 x g for 20 minutes at 4oC.
The supernatant was collected and mixed with 1/9 volume (440 mM final
concentration) of 3.3M Na acetate buffer, pH 6.1, and cold isopropanol was added to
33% (v/v) to precipitate nucleic acids. This solution was mixed and held at -20oC for
at least two hours before centrifugation at 5000 x g for 20 minutes at 4oC. The
supernatant was discarded and the pellet was dissolved in one mL of autoclaved
nanopure water. CsCl was added to a final concentration of 0.4 g/ml. This solution
26
was then centrifuged at 5000 x g for 10 minutes at 4oC to remove any insoluble
material. A discontinuous CsCl gradient was prepared by layering 500 L of solution
A (5.7 M CsCl and 10mM Na-EDTA made with DEPC treated water with pH adjusted
to 7.5 and autoclaved) at the bottom of an ultracentrifuge tube with a 1 mL of solution
B (2.8 M CsCl, 10mM Na-EDTA made with DEPC treated water, pH adjusted to 7.5
and autoclaved) layered on top of it followed by a layer of 500 L of the sample.
Ultracentrifuge tubes were then balanced with solution C (2.4 M CsCl, 10mM NaEDTA made with DEPC treated water and pH adjusted to 7.5 and autoclaved). The
samples were then centrifuged on a Beckman tabletop TL-100 ultracentrifuge in a
TLS-55 rotor at 106,000 x g for 23 hours at 11oC. The supernatant was removed by
pipetting from top to bottom. The pellet containing the RNA was then resuspended in
L of autoclaved nanopure water and centrifuged at 14000 x g for 15 minutes at
4oC (39). This supernatant was then subjected to spectrophotometric analysis and
quantitation.
UV spectroscopy was carried out using an HP 8452 A Diode Array
Spectrophotometer. Prior to analysis, a quartz cuvette was soaked in chromic acid for
ten minutes then rinsed ten times with autoclaved nanopure water to remove any
RNase contamination. A 50-fold dilution of the RNA was made by adding 6 L of
isolated RNA to 294 L of nanopure water. Samples were scanned between 200-400
nm. Ratios of A260-A320/A280-A320 were calculated to determine RNA purity.
27
Quantification of RNA was done using the equation: A260 x 40 g/mL x 50 (dilution
factor).
Additionally, RNA integrity was analyzed by denaturing agarose gel
electrophoresis. Electrophoresis equipment was washed and rinsed in nanopure water,
soaked in 3% H2O2 for 10 minutes and rinsed with DEPC treated water. All solutions
were made with DEPC treated water. RNA samples were separated by gel
electrophoresis using a 1.2% (w/v) formaldehyde denaturing agarose gel (40) and
stained with ethidium bromide.
2.7 Amplification of Monoterpene Synthase Gene Candidates
2.7.1 Degenerate Primers
Degenerate primers were designed based on homologous domains in known
monoterpene synthase protein sequences. Twenty-six monoterpene synthase protein
sequences from the order Pinales were selected from the NCBI Entrez protein
database. Conserved amino acid motifs were identified by the alignment of these
sequences using the ClustalW multiple alignment program (41). These sequences
were subjected to the Block multiple alignment processor program and used in the
consensus-degenerate hybrid oligonucleotide primer (CODEHOP) program (42). This
program designs degenerate primers for use in polymerase chain reactions (PCR).
Three degenerate primers were chosen based on Tm, nucleotide length and clamp
scores (Table 1).
28
Table 2. Degenerate primers designed using CODEHOP. N=A+T+G+C, D=G+A+T,
H=A+T+C, R=A+G, W=A+T and Y=C+T.
Block C forward primer
Block K reverse primer
Block O reverse primer
GAATGGGAGACTACCACTCCAAYHWNTGGRA
TGGTTCCGAAGGTGTCGTANAYRTCRTC
GATCCGGGGTGGTCCTTNADRTARAC
2.7.2 Synthesis of 5’ RACE-ready cDNA and 3’ RACE-ready cDNA
First-strand cDNA for 5’ and 3’ RACE was generated by reverse transcription
of the isolated RNA using the SMARTer™ RACE cDNA Amplification Kit (Clontech)
according to the manufacturer’s instructions. This system uses SMARTScribe RT
(reverse transcriptase), which has terminal transferase activity that adds 3-5 residues at
the 3’ end of the first strand of 5’ cDNA. The SMARTer oligonucleotide contains a
terminal stretch of modified bases that anneals to the extended cDNA, where the
oligonucleotide can then serve as an extended template for the reverse transcriptase
leading to the generation of a complete cDNA copy of the original RNA with the
addition of the extra SMARTer sequence at the end (Figure 9). Because the template
switching occurs only when the reverse transcriptase reaches the end of the RNA
template the SMARTer sequence is usually only incorporated into full-length first
strand cDNAs (43). The first-strand generation of 3’ RACE-ready cDNA utilizes a
modified oligo T primer that contains the SMARTer oligonucleotide sequence which
allows use of the universal primer for subsequent amplification (Figure 10).
29
Figure 9. Mechanism of first-strand SMARTer 5’ cDNA synthesis.
Figure 10. Mechanism of first-strand SMARTer 3’ cDNA synthesis.
2.7.3 Rapid Amplification of cDNA Ends (RACE)
Following the generation of 5’-RACE-Ready cDNA and 3’-RACE-Ready
cDNA, 5’-RACE and 3’-RACE were performed using the SMARTer RACE cDNA
Amplification kit. 5’-RACE-Ready cDNA was used as the template with the universal
primer as the forward primer and either the Block K or Block O degenerate primers as
the reverse primer. Additionally 3’-RACE-Ready cDNA was used as the template
with the forward degenerate primer Block C and the universal primer as the reverse
30
primer, according to the manufacturer’s protocol. PCR was performed on an
Eppendorf Mastercycler® Personal PCR Thermocycler. The PCR program used was:
35 cycles at 94 oC for 30 seconds, 46 oC for 30 seconds, and 72 oC for 3 minutes
followed by 10 minute extension time at 72oC.
Samples of cDNA, synthesized with the SMARTerTM RACE kit, along with a
100 bp ladder (Fisher Scientific) were separated by electrophoresis in a 1.2% (w/v)
agarose/0.005% (w/v) EtBr gel in 22.5 mM Tris-borate (pH8), 0.5mM EDTA buffer.
The gel was run at 85 volts for 2-3 hours.
2.7.4 Gel Purification of PCR Products
PCR products that had been separated by gel electrophoresis that were of
appropriate size were excised and subjected to Nucleo Trap® extraction via
manufacturer’s protocol. These extracts were subjected to gel electrophoresis to
determine their concentration by comparison with the molecular weight markers’
concentrations and then sent for sequencing.
2.7.5 Cloning of Amplified Sequences
Gel-purified or unpurified PCR products were cloned into a
pCR®8/GW/TOPO® vector and then transformed into One Shot® chemically
competent E. coli cells according to the manufacturer’s protocol. These cells were
31
then plated on LB media plates containing 100 g/ml spectinomycin and incubated
overnight at 37oC.
Antibiotic-resistant colonies from the transformation were screened for inserts
by electrophoretic analysis of plasmid size as follows. Colonies were picked using a
sterile pipet tip, streaked on master LB spectinomycin plate (one per colony), and
placed in separate polypropylene tubes containing l of 1X lysis buffer (10% w/v
sucrose, 100 mM NaOH, 60 mM KCl, 5 mM EDTA, 0.25% (w/v) SDS). The tubes
were incubated in a 37 oC water bath for 5 minutes, placed on ice for 5 minutes and
then centrifuged at 14,000 x g for 10 minutes (44). Loading dye (2L) was added to
10 l of each supernatant and then loaded onto a 0.8% agarose gel made with 0.5%
TBE, along with 1 l of a Hind III digest of lambda phage (Lambda Hind III) as a
molecular marker. The gel was run at 100 volts for 1.5 – 2 hours.
Colonies containing plasmids with inserts, as determined by electrophoretic
analysis, were then subjected to PCR screening using the appropriate degenerate
primer and a vector-based primer to confirm that the insert was the PCR product, as
follows. PCR-grade water (5 l) was inoculated with a colony, and 8 l of 5 Prime
Master Mix (5 Prime) was added, along with 0.5 l of appropriate degenerate primer
and 0.5 l of vector-based primer, and 6 l of PCR-grade water was added to bring the
final volume to 20 l (45). The PCR program used was: 94 oC for 5 minutes, followed
by 29 cycles of 94 oC for 30 seconds, 60 oC for 30 seconds, 72 oC for 2 minutes,
32
followed by a 10 minute extension period at 72 oC. The products were then separated
on a 1.2% agarose gel to determine directionality as well as insert size.
2.7.6 Sequencing and Analysis
Colonies that contain plasmids with inserts of the expected size as determined
by PCR screening were then grown overnight in 3 ml of LB media containing 100
g/ml spectinomycin for plasmid isolation. Plasmids were isolated using a BioRad
miniprep kit, according to manufacturer’s protocols. Isolated plasmids were
sequenced at Davis Sequencing on an ABI 3730 sequencer using a either a GW1 or
GW2 primer from the TOPO® Cloning Reaction (Invitrogen).
33
Chapter 3
RESULTS AND DISCUSSION
3.1 Initiation and Maintenance of Callus Cultures
The main was to isolate a nucleotide sequence with homology to a
monoterpene synthase gene involved in the biosynthesis of -thujaplicin. One
approach is to isolate mRNA from tissue that is actively synthesizing -thujaplicin to
obtain cDNA, which can serve as a template for PCR amplification of monoterpene
synthase cDNA using degenerate primers designed based on conserved amino acid
sequences among monoterpene synthases. First tissue needed to be obtained that was
actively producing -thujaplicin from which to obtain mRNA. Because -thujaplicin
is normally found in the heartwood of mature trees from Cupressaceae, and a mature
tree could not be sacrificed to obtain RNA from the heartwood, callus cultures were
initiated and screened for -thujaplicin accumulation. Callus cultures are
undifferentiated cells that have the ability to produce the range of chemicals present in
the parent plant and have been used to produce -thujaplicin (26, 29, 32).
Sections of small branches from six different Cupressaceae species (Cupressus
dupreziana, Cupressus lawsoniana, Cupressus lusitanica, Cupressus macnabiana,
Cupressus nootkatensis and Thuja orientalis) were sampled from local arboretums,
surface sterilized and placed on solid Murashige and Skoog media to induce callus
proliferation. It took three to six weeks for the calli to grow and become established,
although cells from C. macnabiana and T. orientalis did not grow. Growth of the calli
34
were monitored, and those of C. dupreziana grew the largest (Figure 11). These calli
were maintained by replating every 4-6 weeks (Figure 12).
Callus diameter (mm)
25
20
15
10
5
0
Figure 11. Average callus size after six weeks. Values and error bars represent means
and standard errors of seven calli.
Figure 12. C. dupreziana callus cutures.
35
3.2 Analysis of -thujaplicin and Other Monoterpene Content in Callus Cultures
To determine whether the established calli produce -thujaplicin GC-MS
analysis was employed. Initial GC-MS analysis of underivatized -thujaplicin
resulted in very poor peak shape. Compounds that are not volatile or lack thermal
stability can be derivatized to allow for analysis via GC-MS, so derivatization with
BSTFA and 1% TMCS was used to increase the volatility and stability of -thujaplicin
to allow for better peak shape and easier detection and quantification by GC-MS.
Derivatization modifies the polar OH group on -thujaplicin by replacing the
hydrogen with a trimethylsilyl group allowing for a less polar and more volatile
compound (Figure 13).
O
CH3
O
OH
O
BSFTA + 1% TMCS
Si
CH3
CH3
Figure 13. Derivatization of -thujaplicin
A derivatized sample of -thujaplicin standard with an internal standard, 1,3napthalenediol, for quantification which was used in all extractions, was subjected to
36
GC-MS analysis for comparison with derivatized callus extracts (Figure 14).
Derivatized -thujaplicin has a molecular weight of 236.3, and the mass ion can be
detected in the mass spectrum (Figure 15). The mass spectrum also has a base ion
peak of m/z 221.3, likely due to the loss of a methyl group from the trimethyl silyl
group. This is the first reported mass spectrum of trimethylsilyl -thujaplicin.
3000000
-thujaplicin
2500000
Abundance
2000000
1500000
1,3-napthalenediol
1000000
500000
0
10.5
11
11.5
12
12.5
13
13.5
14
Retention Time (min)
Figure 14. Total ion chromatogram of derivatized 1.0 mg/mL -thujaplicin with 1.0
mg/mL 1,3-napthalenediol as the internal standard.
14.5
37
A b u n d a n c e
S c a n
1 8 0
( 1 2 .0 2 6
m in ) : 0 2 0 9 0 9 C .D \ d a ta .m s
2 2 1 .3
3 4 0 0 0 0
3 2 0 0 0 0
3 0 0 0 0 0
2 8 0 0 0 0
2 6 0 0 0 0
A
b
u
n
d
a
n
ce
2 4 0 0 0 0
2 2 0 0 0 0
2 0 0 0 0 0
1 8 0 0 0 0
1 6 0 0 0 0
1 4 0 0 0 0
1 2 0 0 0 0
1 0 0 0 0 0
2 0 6 .3
8 0 0 0 0
7 3 .2
6 0 0 0 0
4 0 0 0 0
1 7 9 .3
9 1 .2
2 0 0 0 0
1 3 5 .2
5 1 .0
1 5 1 .2
1 1 5 .2
2 3 6 .3
0
4 0
m /z -->
5 0
6 0
7 0
8 0
9 0
1 0 0
1 1 0
1 2 0
1 3 0
1 4 0
1 5 0
1 6 0
1 7 0
1 8 0
1 9 0
2 0 0
2 1 0
2 2 0
2 3 0
2 4 0
m/z
Figure 15. Mass spectrum ofderivatized -thujaplicin standard.
A callus from each established species was then extracted with ethyl acetate
containing the internal standard and analyzed by GC-MS after derivatization with
BSFTA-TMCS. Total ion chromatograms were obtained for derivatized extracts from
each callus to determine which species produced the most -thujaplicin.
Quantification of -thujaplicin was achieved by comparision of the peak areas to that
of the internal standard. Calli from two of the four species analyzed (C. dupreziana
and C. lusitanica) had detectable -thujaplicin, based on a similar retention times and
mass spectra as the -thujaplicin standard (Figure 16 and 17). To establish the
variability of -thujaplicin production for each of these two species, five calli from C.
lusitanica and eight from C. dupreziana were analyzed. -thujaplicin was detected in
38
3000000
2500000
-thujaplicin
Abundance
2000000
1500000
1000000
1,3-napthalenediol
500000
0
8.5
9
9.5
10 10.5 11 11.5 12 12.5 13 13.5 14 14.5 15 15.5 16 16.5 17 17.5 18
Retention Time (min)
400000
350000
1,3-napthalenediol
Abundance
300000
250000
200000
150000
-thujaplicin
100000
50000
0
8.5
9
9.5
10 10.5 11 11.5 12 12.5 13 13.5 14 14.5 15 15.5 16 16.5 17 17.5 18
Retention Time (min)
Figure 16. Total ion chromatograms of derviatized -thujaplicin and 1,3
napthalenediol standards (top panel) and derivatized extract of C. dupreziana callus
(bottom panel).
39
A b u n d a n c e
S c a n
1 8 0
( 1 2 .0 2 6
m in ) : 0 2 0 9 0 9 C .D \ d a ta .m s
2 2 1 .3
3 4 0 0 0 0
3 2 0 0 0 0
3 0 0 0 0 0
2 8 0 0 0 0
2 6 0 0 0 0
2 4 0 0 0 0
A
bu
nd
an
ce
2 2 0 0 0 0
2 0 0 0 0 0
1 8 0 0 0 0
1 6 0 0 0 0
1 4 0 0 0 0
1 2 0 0 0 0
1 0 0 0 0 0
2 0 6 .3
8 0 0 0 0
7 3 .2
6 0 0 0 0
4 0 0 0 0
1 7 9 .3
9 1 .2
2 0 0 0 0
1 3 5 .2
5 1 .0
1 5 1 .2
1 1 5 .2
2 3 6 .3
0
4 0
5 0
6 0
7 0
8 0
9 0
1 0 0
1 1 0
1 2 0
m /z -->
1 3 0
1 4 0
1 5 0
1 6 0
1 7 0
1 8 0
1 9 0
2 0 0
2 1 0
2 2 0
2 3 0
2 4 0
m/z
A b u n d a n c e
S c a n 1 7 3 ( 1 1 .8 7 4 m in ) : 0 4 2 4 0 9 A .D \ d a ta .m s
2 2 1 .2
1 6 0 0 0
1 5 0 0 0
1 4 0 0 0
1 3 0 0 0
1 2 0 0 0
1 1 0 0 0
A
1 0 0 0 0
bu
9 0 0 0
nd
8 0 0 0
an 7 0 0 0
ce 6 0 0 0
7 3 .0
5 0 0 0
2 0 6 .2
4 0 0 0
3 0 0 0
2 0 0 0
9 1 .1
5 5 .0
1 7 9 .2
1 5 1 .1
1 3 5 .1
1 0 0 0
1 1 7 .1
2 3 6 .2
0
4 0
5 0
6 0
7 0
8 0
9 0
1 0 0
1 1 0
1 2 0
1 3 0
1 4 0
1 5 0
1 6 0
1 7 0
1 8 0
1 9 0
2 0 0
2 1 0
2 2 0
2 3 0
2 4 0
m /z -->
m/z
Figure 17. Mass spectrum of -thujaplicin standard after derivatization (top panel) and
derivatized -thujaplicin from extracts of C. dupreziana (lower panel).
40
five of the eight C. dupreziana calli tested, whereas -thujaplicin was detected in only
one C. lusitanica callus. The C. dupreziana calli in which -thujaplicin was detected
had average levels of -thujaplicin of 0.01 mg per gram of callus similar to levels
found in related C.lusitanica calli in other studies.
From these results, it was determined that C. dupreziana calli more
consistently produced
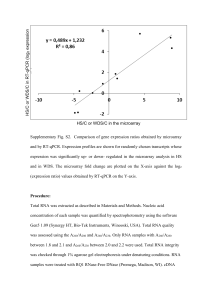
, as well as having the fastest calli
growth. Although other groups have used C. lusitanica cultures as an experimental
system for studying -thujaplicin production (26, 29, 46), C. dupreziana cells were
chosen because of the more consistent production of -thujaplicin.
To obtain a more complete understanding of the monoterpene composition
within C. dupreziana callus cultures, non-thujaplicin monoterpenes were extracted by
steam distillation of calli and analyzed by GC-MS. The installation of a new column
on the GC-MS allowed for analysis of both non-thujaplicin monoterpenes and thujaplicin with good peak shape without derivatization. Based on the mass spectra
and retention times, some of the monoterpenes detected in C. dupreziana callus
cultures were : -pinene, 3-carene, limonene, terpinolene, carvacrol and -thujaplicin
(Figure 18). C. lusitanica callus cultures also contain -pinene, limonene and
terpinolene; however, other monoterpenes present in C. lusitanica calli that we did not
detect in C. dupreziana include -ocimene, myrcene and sabinene.
41
600000
6
500000
400000
Abundance
300000
200000
1
100000
2
3
0
7
8
9
10
5
4
11
12
13
14
15
16
17
18
19
20
21
22
Retention Time (min)
Figure 18. Total ion chromatogram of C. dupreziana callus culture (1) -pinene, (2) 3carene, (3) limonene, (4) terpinolene, (5) carvacrol and (6) -thujaplicin.
3.3 Elicitation of -thujaplicin by Methyl Jasmonate
-thujaplicin biosynthesis has been shown to be elicited by methyl jasmonate,
a compound similar to -thujaplicin that is involved in plant defense responses, or
yeast elicitor (26-28, 46, 47). We tested the effects of methyl jasmonate on four C.
dupreziana calli. After treatment with 200 M methyl jasmonate for one week, calli
showed a two- to three-fold increase in-thujaplicin production compared to the
control calli. The elicited calli had an average 0.030 mg -thujaplicin per gram of
23
42
callus while the unelicited calli had an average of 0.0133 mg of -thujaplicin per gram
of callus. This three-fold increase in -thujaplicin production is less than the 10-fold
increase seen after the addition of fungal elicitors to C. lusitanica callus cultures and
the ten-fold increase in -thujaplicin production after the addition of 200 M methyl
jasomate to C. lusitanica suspension cultures (26, 34, 46).
Because calli treated with methyl jasmonate had increased -thujaplicin
production they were promising candidates for RNA isolation. However mold formed
on the calli preventing their use as an RNA source. At the time we did not have
enough calli and so after several unsuccessful attempts to isolate RNA from elicited
calli we isolated RNA from unelicited calli which still produced significant levels of
-thujaplicin but were less prone to mold development.
3.4 Isolation of RNA from C. dupreziana Callus Cultures
Having developed a tissue culture system that produces significant levels of thujaplicin, RNA isolation to generate a template for isolation of monoterpene
synthase cDNAs was then pursued. RNA isolation from callus cultures is very
difficult due to the high concentration of soluble polysaccharides and polyphenols
(39). Phenolic substances can bind and inactivate RNA, and polysaccharides can coprecipitate with RNA and interfere with subsequent isolation steps (39, 48). Six
different RNA extraction methods, including two commercial plant RNA extraction
kits, were evaluated for their ability to yield RNA of sufficient quantity and quality for
43
cDNA synthesis. A version of the Lewinsohn protocol (39), slightly modified by the
absence of insoluble PVPP as well as being scaled down, yielded viable RNA. This
method employs polyvinylpyrrolidone (PVP) to bind phenolic compounds, EDTA and
thiourea to inhibit polyphenoloxidases, and aurintricarboxylic acid to inhibit RNases.
The method also includes a 10% (v/v) ethanol precipitation to remove non-nucleic
acid associated polysaccharides.
The purity and concentration of RNA was evaluated by UV-VIS
spectrophotometry. Purity of RNA is crucial for RT-PCR cDNA synthesis. Spectra in
the range of 200-400 nm were obtained and readings at A260, A280 and A320 were taken.
By using the equation: A260-A320/A280-A320, an assessment of RNA purity can be made.
Pure RNA absorbs maximally at 260 nm whereas protein, a source of contamination,
absorbs maximally at 280 nm. The absorbance at 320 nm is a measure of background
absorbance, turbidity and scatter, and by subtracting this from A260 and A280 values a
more accurate estimation of RNA purity can be obtained. If the ratio is greater than
~1.8 it is indicative of pure RNA. Furthermore, an approximate quantification of RNA
can also be made by using the equation: concentration of RNA = A260 x 40
RNA/mL x dilution factor. RNA from two C. dupreziana calli, A and B, were isolated
and analyzed using UV-VIS spectrophotometry. A260-A320/A280-A320 ratios of 1.77 and
1.74 were obtained, and concentrations of 194g/mL and 828.7g/mL were calculated
for RNA isolated from calli A and B, respectively. The ratios were slightly lower than
44
desired but the UV spectra indicated the RNA was of sufficient quality for cDNA
synthesis (Figure 19).
Figure 19. UV-VIS spectrum of isolated RNA from callus B.
To insure the isolated RNA was not significantly degraded, a sample of the
isolated RNA was subjected to agarose gel electrophoretic analysis. Unlike DNA,
RNA is single-stranded and, thus, able to form secondary structures via intramolecular
base pairing which can affect its electrophoretic mobility. Consequently, RNA was
electrophoresed under denaturing conditions to disrupt RNA secondary structure.
After electrophoresis on a denaturing gel, ribosomal bands should be visible; if not,
the RNA is not intact. An agarose formaldehyde gel revealed the presence of distinct,
albeit faint, ribosomal bands for RNA from calli A and B, similar to that found in
preparations of high quality RNA from wheat grass (Figure 20).
45
1
2
3
Figure 20. Denaturing electrophoretic agarose (1.0 %) gel of RNA extracted from
calli. Lane 1: wheat grass RNA control, Lane 2 : callus A, Lane 3 : callus B (From left
to right).
To confirm that the calli from which RNA was isolated were actively
accumulating -thujaplicin, half of each callus used for RNA isolation was extracted
with ethyl acetate containing 2-napthol as an internal standard and analyzed by GCMS. A -thujaplicin standard with 2-napthol was subjected to GC-MS analysis as a
control (Figure 21). -thujaplicin has a molecular weight of 164.1, and the mass ion
can be detected in the mass spectrum, which is similar to a previously reported (24) thujaplicin mass spectrum (Figure 22). This control was used for comparision with
extracted calli after RNA isolation. In both callus A and callus B, -thujaplicin was
46
detected by GC-MS (Figure 23) with an average of 1.3 g of -thujaplicin per gram of
callus.
300000
250000
-thujaplicin
Abundance
200000
150000
100000
2-napthol
50000
0
6
6.5
7
7.5
8
8.5
9
9.5
10
Retention Time (min)
10.5
11
11.5
Figure 21. Total ion chromatogram of 1.0 mg/mL -thujaplicin with 0.1 mg/mL 2A b und a nc e
napthol
as the internal standard.
S c a n 1 0 1 1 (8 .4 8 5 m in ): B T H U J A S T N D .D \ d a ta .m s
1 2 1 .1
55000
50000
45000
40000
Ab
un
da
nc
e
35000
30000
1 6 4 .1
25000
20000
15000
7 7 .1
9 1 .1
10000
5000
1 0 3 .1
6 5 .1
4 1 .1
5 1 .1
40
50
1 3 6 .1
1 4 9 .1
0
60
70
80
90
100
110
m / z -->
m/z
Figure 22. Mass spectrum of -thujaplicin standard.
120
130
140
150
160
170
12
47
300000
250000
200000
Abundance
-thujaplicin
150000
100000
2-napthol
50000
0
8
8.1 8.2 8.3 8.4 8.5 8.6 8.7 8.8 8.9
9
9.1 9.2 9.3 9.4 9.5
600000
500000
Abundance
400000
2-napthol
300000
200000
-thujaplicin
100000
0
8
8.1 8.2 8.3 8.4 8.5 8.6 8.7 8.8 8.9
9
9.1 9.2 9.3 9.4 9.5
Retention time (min)
Figure 23. Total ion chromatograms of -thujaplicin and 2-napthol standards (upper
panel) and C. dupreziana callus B extract with 0.025 mg/mL 2-napthol as internal
standard (lower panel).
48
3.5 Degenerate Primer Design
Because the goal was to identify a monoterpene synthase cDNA sequence and
the exact sequence was unknown, degenerate primers were employed. Degenerate
primers are used to amplify unknown DNA sequences when the protein sequence is
known or when protein sequences from other species are known. Because the genetic
code is degenerate, i.e., more than one codon can specify the same amino acid (Table
3), degenerate primers contain a pool of all possible combinations of nucleotides that
code for a given amino acid sequence. Often protein sequences are conserved
between different species, whereas DNA sequences are not, so degenerate primers
designed based on conserved amino acid sequences can allow amplification of
nucleotides encoding the conserved protein. This approach has been used in other
studies to identify monoterpene synthase genes in novel organisms (21, 35, 49-53).
Table 3. Codon degeneracy
Number of codons
1
2
3
4
6
Amino Acids
Met, Trp
Phe, Tyr, His, Gln, Asn, Lys, Asp, Glu, Cys
Ile
Val, Pro, Thr, Ala, Gly
Leu, Ser, Arg
A search of the NCBI non-redundant protein sequence database revealed 183
monoterpene synthase sequences from many different species, including Abies grandis
and Pinus taeda. Twenty-six monoterpene synthase sequences from conifer species
were aligned using the multiple alignment sequencing program ClustalW to identify
49
conserved domains within these sequences. Monoterpene synthases have two wellknown conserved motifs with known function (49, 54), the RRX8W motif commonly
found in cyclizing monoterpene synthases and DDXXD motif critical in divalent
cation-assisted substrate binding, which were both present in the alignments. In
Figure 24, portions of the aligned sequences are shown with the RRX8W and DDXXD
motifs are highlighted.
Block C
gi|62511233|sp|Q9M7C9.1|TPSDB_
gi|7381253|gb|AAF61455.1|AF139
gi|17367918|sp|O22340.1|TPSDA_
gi|2429145|gb|AAB70907.1|
gi|15080741|gb|AAK83565.1|AF32
gi|17367924|sp|O24475.1|TPSD3_
gi|2411483|gb|AAB71085.1|
gi|15080739|gb|AAK83564.1|
gi|28894482|gb|AAO61225.1|
gi|34582667|gb|AAP72020.1|
gi|28894486|gb|AAO61227.1|
gi|62511221|sp|Q948Z0.1|TPSD6_
gi|2411485|gb|AAB70707.1|
gi|28894488|gb|AAO61228.1|
gi|28894490|gb|AAO61229.1|
gi|62511234|sp|Q9M7D0.1|TPSD9_
gi|7381251|gb|AAF61454.1|AF139
gi|77454875|gb|ABA86247.1|
gi|62511235|sp|Q9M7D1.1|TPSD8_
gi|7381249|gb|AAF61453.1|AF139
gi|77454877|gb|ABA86248.1|
gi|21322150|gb|AAK39127.2|
gi|2411481|gb|AAB71084.1|
gi|17367921|sp|O24474.1|TPSD2_
gi|21322152|gb|AAK39128.2|AF36
gi|35764438|dbj|BAC92722.1|
V---INMKLTTVSHRDDNGGGVLQRRIADHHPNLWEDDFIQSLS-SPYGG
V---INMKLTTVSHRDDNGGGVLQRRIADHHPNLWEDDFIQSLS-SPYGG
V---INMKLTTVSHRDDNGGGVLQRRIADHHPNLWEDDFIQSLS-SPYGG
V---INMKLTTVSHRDDNGGGVLQRRIADHHPNLWEDDFIQSLS-SPYGG
V---INMKLTTVSHRDDNDGGVLQRRIADHHPNLWEDDFIQSLS-SPYGG
T-PSISMSSTTVVTDD-----GVRRRMGDFHSNLWDDDVIQSLP-TAYEE
T-PSISMSSTTVVTDD-----GVRRRMGDFHSNLWDDDVIQSLP-TAYEE
T-PSISMSSTTVVTDD-----GVRRRMGDFHSNLWDDDVIQSLP-TAYEE
TRASMSMNLRTAVSDD-----AVIRRRGDFHSNLWDDDLIQSLS-SPYGE
T-PSMSMSLTTTVSDD-----GVQRRMGDFHSNLWNDDFIQSLS-TSYGE
R---PSMSLSTVASED-----DIQRRTGGYLSNLWNDDVIQFLS-TPYGE
A-HSINMCLTSVASTD-----SVQRRVGNYHSNLWDDDFIQSLISTPYGA
A-HSINMCLTSVASTD-----SVQRRVGNYHSNLWDDDFIQSLISTPYGA
A-PSMSMSSTTSVSNED----GVPRRIAGHHSNLWDDDSIASLS-TSYEA
R-PSIRVSSTASVSNDD----GVRRRVGDYRYNHWDEDLIDSLA-TSYEA
T-HSLRMSLSTAVSDDH----GVQRRIVEFHSNLWDDDFIQSLS-TPYGA
T-HSLRMSLSTAVSDDH----GVQRRIVEFHSNLWDDDFIQSLS-TPYGA
T-PSISMCWTATVLDD-----GVQRRIANHHSNLWDDSFIQSLS-TPYGE
TP-SVSMSLTTAVSDDG-----LQRRIGDYHSNLWDDDFIQSLS-TPYGE
TP-SVSMSLTTAVSDDG-----LQRRIGDYHSNLWDDDFIQSLS-TPYGE
APASMSMILTAAVSDDD----RVQRRRGNYHSNLWDDDFIQSLS-TPYGE
APASTSMILTAAVSDDD----RVQRRRGNYHSNLWDDDFIQSLS-TPYGE
TP-SMSISLATAAPD-D----GVQRRIGDYHSNIWDDDFIQSLS-TPYGE
TP-SMSISLATAAPD-D----GVQRRIGDYHSNIWDDDFIQSLS-TPYGE
TP-SMSMSLNTVVSDND----AVQRRIGDYHSNLWNDDFIQSLT-TPYGA
R---NTMAMATTSVES------VTRRTGNHHGNLWDDDFIQSLPKLPYDA
:
:
: **
. * *::. * *
.*
94
94
94
94
94
88
88
83
90
88
88
84
84
89
88
89
89
87
86
86
90
90
88
88
89
79
50
Block K
gi|62511233|sp|Q9M7C9.1|TPSDB_
gi|7381253|gb|AAF61455.1|AF139
gi|17367918|sp|O22340.1|TPSDA_
gi|2429145|gb|AAB70907.1|
gi|15080741|gb|AAK83565.1|AF32
gi|17367924|sp|O24475.1|TPSD3_
gi|2411483|gb|AAB71085.1|
gi|15080739|gb|AAK83564.1|
gi|28894482|gb|AAO61225.1|
gi|34582667|gb|AAP72020.1|
gi|28894486|gb|AAO61227.1|
gi|62511221|sp|Q948Z0.1|TPSD6_
gi|2411485|gb|AAB70707.1|
gi|28894488|gb|AAO61228.1|
gi|28894490|gb|AAO61229.1|
gi|62511234|sp|Q9M7D0.1|TPSD9_
gi|7381251|gb|AAF61454.1|AF139
gi|77454875|gb|ABA86247.1|
gi|62511235|sp|Q9M7D1.1|TPSD8_
gi|7381249|gb|AAF61453.1|AF139
gi|77454877|gb|ABA86248.1|
gi|21322150|gb|AAK39127.2|
gi|2411481|gb|AAB71084.1|
gi|17367921|sp|O24474.1|TPSD2_
gi|21322152|gb|AAK39128.2|AF36
gi|35764438|dbj|BAC92722.1|
gi|62511233|sp|Q9M7C9.1|TPSDB_
gi|7381253|gb|AAF61455.1|AF139
gi|17367918|sp|O22340.1|TPSDA_
gi|2429145|gb|AAB70907.1|
gi|15080741|gb|AAK83565.1|AF32
gi|62511234|sp|Q9M7D0.1|TPSD9_
gi|7381251|gb|AAF61454.1|AF139
gi|77454875|gb|ABA86247.1|
gi|28894488|gb|AAO61228.1|
gi|28894490|gb|AAO61229.1|
gi|17367924|sp|O24475.1|TPSD3_
gi|2411483|gb|AAB71085.1|
gi|15080739|gb|AAK83564.1|
gi|34582667|gb|AAP72020.1|
gi|28894482|gb|AAO61225.1|
gi|28894486|gb|AAO61227.1|
gi|62511221|sp|Q948Z0.1|TPSD6_
gi|2411485|gb|AAB70707.1|
gi|62511235|sp|Q9M7D1.1|TPSD8_
gi|7381249|gb|AAF61453.1|AF139
gi|77454877|gb|ABA86248.1|
gi|21322150|gb|AAK39127.2|
gi|2411481|gb|AAB71084.1|
gi|17367921|sp|O24474.1|TPSD2_
gi|21322152|gb|AAK39128.2|AF36
gi|35764438|dbj|BAC92722.1|
LLTVLDDMYDTFGTLDELQLFTTAFKRWDLSETKCLPEYMKAVYMDLYQC
LLTVLDDMYDTFGTLDELQLFTTAFKRWDLSETKCLPEYMKAVYMDLYQC
LVTVLDDIYDTFGTMNELQLFTDAIKRWDLSTTRWLPEYMKGVYMDLYQC
LVTVLDDIYDTFGTMNELQLFTDAIKRWDLSTTRWLPEYMKGVYMDLYQC
LVTVLDDIYDTFGTMNELQLFTDAIKRWDLSTTRWLPEYMKGVYMDLYQC
LITVLDDMYDTFGTVDELELFTATMKRWDPSSIDCLPEYMKGVYIAVYDT
LITVLDDMYDTFGTVDELELFTATMKRWDPSSIDCLPEYMKGVYIAVYDT
LITVLDDMYDTFGTVDELELFTATMKRWDPSSIDCLPEYMKGVYIAVYDT
IITVLDDMYDTFGTLDELELFTSAIKRWDPSATECLPEYMKGVYMIVYNT
IITILDDMYDTFGTVDELELFTAAMKRWDPSAADCLPEYMKGVYLILYDT
ILTVLDDMYDLFGTVDELKLFTAAIKRWDPSATDCLPQYMKGIYMMVYNT
LITVLDDMYDVFGTVDELELFTATIKRWDPSAMECLPEYMKGVYMMVYHT
LITVLDDMYDVFGTVDELELFTATIKRWDPSAMECLPEYMKGVYMMVYHT
MITILDDIYDTFGTMEELKLLTAAFKRWDPSSIECLPDYMKGVYMAVYDN
FVTVLDDIYDTYGTMEELELFTAAIKRWDPSVVDCLPEYMKGVYMAVYDT
LITVLDDIYDTFGTMEELELFTAAFKRWDPSATDLLPEYMKGLYMVVYET
LITVLDDIYDTFGTMEELELFTAAFKRWDPSATDLLPEYMKGLYMVVYET
LGTVLDDIYDTFGTMDELELFTAAVKRWHPSAAEGLPEYMKGVYMMFYET
LITVLDDIYDTFGTMDEIELFNEAVRRWNPSEKERLPEYMKEIYMALYEA
LITVLDDIYDTFGTMDEIELFNEAVRRWNPSEKERLPEYMKEIYMALYEA
LNTVLDDIYDTFGTMDEIELFTEAVRRWDPSETESLPDYMKGVYMVLYEA
LNTVLDDIYDTFGTMDEIELFTEAVRRWDPSETESLPDYMKGVYMVLYEA
LVTVLDDIYDTFGTIDELELFTSAIKRWNSSEIEHLPEYMKCVYMVVFET
LVTVLDDIYDTFGTIDELELFTSAIKRWNSSEIEHLPEYMKCVYMVVFET
LVTVLDDVYDTFGKMDELELFTAAVKRWDLSETERLPEYMKGLYVVLFET
LATIIDDIYDTYGSIEELKHFTEVFKRWDSSPPDYLPEYMKIAYSALYDG
: *::**:** :*.::*:: :. ..:**. *
**:*** * .:.
Block O
DTRCYKADRARGEEASAISCYMKDHPGSTEEDALNHINVMISDAIRELNW
DTRCYKADRARGEEASAISCYMKDHPGSTEEDALNHINVMISDAIRELNW
DTRCYKADRARGEEASAISCYMKDHPGSIEEDALNHINAMISDAIRELNW
DTRCYKADRARGEEASAISCYMKDHPGSIEEDALNHINAMISDAIRELNW
DTRCYKADRARGEEASAISCYMKDHPGSTEEDALNHINAMISDAIRELNW
DTRCYKADRARGEEASCISCYMKENPGSTEEDAINHINAMVNNLIKEVNW
DTRCYKADRARGEEASCISCYMKENPGSTEEDAINHINAMVNNLIKEVNW
DIHSYQAERSRGEESSGISCYMKDNPESTEEDAVTHINAMINRLLKELNW
DTQCYKADRARGEEASAVSCYMKDHPGITEEDAVNQVNAMVDNLTKELNW
------GQGSVVQEASSVSCYMKDNAGLTEEDAIHCINDMVNNLLKELNW
DTRCYKADRARGEEASSISCYMKDNPGVSEEDALDHINAMISDVIKGLNW
DTRCYKADRARGEEASSISCYMKDNPGVSEEDALDHINAMISDVIKGLNW
DTRCYKADRARGEEASSISCYMKDNPGVSEEDALDHINAMISDVIKGLNW
DTRCYKADRARGEEASSISCYMKDNPGATEEDALDHINAMISDVIRGLNW
DTRCYQADRARGEEASCISCYMKDNPGTTEEDALNHLNAMISDVIKGLNW
DTRCYQADRARGEETSCISCYMKDNPGATEEDALNHLNVMISGVIKELNW
DTRCYKADRARGEEASSISCYMKDNPGLTEEDALNHINFMIRDAIRELNW
DTRCYKADRARGEEASSISCYMKDNPGLTEEDALNHINFMIRDAIRELNW
DTRCYKADRDRGEEASSISCYMKDNPGLTEEDALNHINAMINDIIKELNW
DTRCYKADRDRGEEASSISCYMKDNPGLTEEDALNHINAMINDIIKELNW
DTRCYKADRARGEEASCISCYMKDNPGSTEEDALNHINSMINEIIKELNW
DTRCYKADRARGEEASCISCYMKDNPGSTGEDALNHINSMINEIIKELNW
DTRCYKADRDRGEEASCISCYMKDNPGSTEEDALNHINAMVNDIIKELNW
DTRCYKADRDRGEEASCISCYMKDNPGSTEEDALNHINAMVNDIIKELNW
DTRCYXADRARGEEASCISCYMKDHPGSTEEDAVNHINAMINDIIRELNW
DTRTFKAEANRGEVTSSIACYLKEHPESTEKDALKYLQFMLDENLKELNL
.:
: :* ::**:*::.
:**: :: *:
: :*
432
432
432
432
432
423
423
418
424
422
422
413
413
423
416
425
425
421
425
425
429
429
422
422
428
402
582
582
582
582
582
575
575
571
573
560
573
573
568
572
574
572
563
563
575
575
579
579
572
572
578
551
Figure 24. Sections of the aligned monoterpene synthase sequences with RRX8W and
DDXXD motifs highlighted and the primer blocks marked above the sequences.
51
These aligned sequences were then organized into blocks (short multiply aligned
ungapped segments that correspond to highly conserved regions of protein) using the
Blockmaker component of the CODEHOP program (42). The CODEHOP program
then designs degenerate primers for the blocks. Many primers were generated by the
CODEHOP program, and we focused on primers from blocks that are most conserved
among monoterpene synthases. Three primers were chosen from the possible
degenerate primers generated from the CODEHOP program (Figure 24). The primer
from Block C contains a portion of the RRX8W motif whereas Block K contains a
portion of the DDXXD motif, both of which are present in monoterpene synthases.
Block O was chosen from another downstream conserved region. These primers were
then used in 5’ and 3’ RACE PCR.
Table 4. Degenerate primers for amplification of monoterpene symthase cDNA.
N=A+T+G+C, D=G+A+T, H=A+T+C, R=A+G, W=A+T and Y=C+T.
Block C forward primer
Block K reverse primer
Block O reverse primer
GAATGGGAGACTACCACTCCAAYHWNTGGRA
TGGTTCCGAAGGTGTCGTANAYRTCRTC
GATCCGGGGTGGTCCTTNADRTARAC
3.6 Rapid Amplification of cDNA Ends (RACE)
The first step in RACE involves the conversion of RNA to the first strand of
cDNA by use of reverse transcriptase in a manner that allows subsequent
amplification of either the 5’ or 3’ ends of the transcript (Figure 25). The first strand
52
of cDNA that can serve as a template for isolating the 5’ end of the cDNA is known as
5’ RACE-ready cDNA, and the first stand of cDNA that can serve as a template for
isolating the 3’ end of the cDNA is known as 3’ RACE-ready cDNA. In generating 5’
RACE-ready cDNA, the specific system used for this study employs a reverse
transcriptase with terminal transferase activity that adds 3-5 residues to the 3’ end of
the first strand. An oligonucleotide (SMARTer oligo) anneals to the extra bases
allowing for full-length, first-strand cDNA. In generating 3’ RACE-ready cDNA, the
first-strand cDNA is synthesized using a standard reverse transcription procedure but a
special oligo(dT) primer that includes part of the SMARTer oligo sequence at its 5’
end. The SMARTer oligo contains a universal priming sequence for use in the 3’ and
5’ RACE reactions (Figure 25). Once first-strand synthesis is complete, the respective
5’ and 3’ RACE-ready cDNA is used in 5’ and 3’ RACE PCR reactions utilizing genespecific primers and the universal primer that recognizes the SMARTer oligo
sequence.
53
5’ RACE
3’ RACE
3’
RACE
Ready
cDNA
First-strand
synthesis
First-strand
synthesis
5’
RACEReady
cDNA
3’ RACE
5’ RACE
Figure 25. Overview of 5' and 3' first-strand cDNA synthesis and 5' and 3' RACE
PCR.
In our 3’ RACE reaction, the degenerate primer Block C is used as the forward
primer, and the universal primer is used as the reverse primer. In the 5’ RACE
reaction, the universal primer is used as the forward primer, and either degenerate
primers Block K or Block O are used as the reverse primers. Based on the location of
the blocks within monoterpene synthase sequences, the expected products were
54
approximately 1700 base pairs for 3’ RACE with Block C, 1200 base pairs for 5’
RACE with Block K and 1700 base pairs for 5’ RACE with Block O. Initial attempts
of 3’ and 5’ RACE PCR at high annealing temperature of 66 oC were unsuccessful,
but amplification was observed when the annealing temperature was lowered to 48 oC
(Figure 26). Only 5’ RACE PCR using Block K or Block O was successful.
bp
11
1
2
3
4
5
6
7
8
9
10
2686
2000
1500
1200
1000
900
800
700
600
500
Figure 26. Agarose gel electrophoresis of 3’ and 5’ RACE products. Lane 1: 100 bp
ladder (Invitrogen), Lane 2-4: 3’ RACE with Block C, Lane 5-7: 5’ RACE with Block
O primer, Lanes 8-10: 5’ RACE with Block K primer, Lane 11: 100bp ladder
(Invitrogen).
The PCR products from several reactions were combined, and the major PCR
products were gel-purified (Figure 27). The purified products were then submitted for
sequencing using the universal primer. Only the PCR product amplified with Block K
primer yielded a sequence of approximately 400 base pairs.
55
bp
1
2
3
4
23130
9416
6557
4361
2322
2027
2686
2000
1500
1200
1000
900
800
700
600
500
564
Figure 27. Agarose gel electrophoresis of 5’ RACE products. Lane 1: Lamda III Hind
digest (Invitrogen), Lane 2: Callus B 5' RACE with Block O primer; boxed band
excised (~1500 base pairs), Lane 3: 100 base pair ladder (Fisher Scientific), Lane 4:
Callus B 5’ RACE with Block K primer; boxed band excised (~850 base pairs).
Homology searches for all sequences were performed with BLASTx and
tBLASTx programs (55). BLASTx translates a nucleotide sequence into all six
reading frames and compares the results to a protein sequence database whereas
tBLASTx translates a nucleotide sequence into all six reading frames and compares
them to translations of a nucleotide sequence database. Based on BLASTx and
tBLASTx homology searches of the NCBI non-redundant protein and nucleotide
sequence databases, this sequence was not homologous to any previously described
monoterpene synthase sequences. However, the 5’ end of the cDNA is likely to
56
contain an unconserved 5’ untranslated region and the 5’ end of the coding region is
likely to encode a poorly conserved plastid targeting sequence (23).
Two non-degenerate gene specific primers (Table 5) were designed from the
obtained sequence to perform 3’ RACE to obtain a full-length copy of cDNA
sequence. Because non-degenerate primers were used the annealing temperature was
raised from 48 oC to 66 oC. Amplification with GSP 1 was unsuccessful.
Amplification with GSP 2 yielded a ~ 1800 bp fragment and a ~ 3000 bp fragment
(Figure 28).
Table 5. Gene specific primers.
Gene Specific Primer 1 (GSP 1)
Gene Specific Primer 2 (GSP 2)
AGGGAGATTTCTAGTGGCAGGC
TTCAAGGGGTGTGGCCACT
57
bp
1
2
3
2686
2000
1500
1200
1000
900
800
700
600
Figure 28. Lane 1: 100 base pair ladder (Fisher Scientific), Lane 2: Callus B 3’ RACE
with GSP 1, Lane 3: Callus B 3’ RACE with GSP 2 (excised bands boxed).
These fragments were excised and purified. Attempts to directly sequence were
unsuccessful, probably due to low concentration of DNA template, so another round
of 3’ RACE with gene specific primer was performed using the same PCR program
(Figure 29) and the resultant PCR products were cloned into a pCR®8/GW/TOPO®
vector and then transformed into One Shot® chemically competent E. coli cells. The
vector contains primer binding sequences GW1 and GW2 flanking the 5’ and 3’ edges
of the cloning site which allows for amplification and sequencing with minimal
amount of vector-encoded DNA. The vector also contains a spectinomycin resistant
gene for selection. The cells were plated and grown overnight on 100 g/mL
spectinomycin LB plates.
58
bp
1
2
2686
2000
1500
1200
1000
900
800
700
600
500
Figure 29. Lane 1: 100 base pair ladder (Fisher Scientific) Lane 2: Fresh 3' RACE
PCR product using GSP 2.
To identify colonies containing plasmids with inserts, samples of 36 colonies
were lysed, and the plasmid size was determined by electrophoresis (Figure 30). The
pCR®8/GW/TOPO® vector without insert has a size of 2817 bp. When colonies are
lysed and the contents run on an agarose gel, plasmids that are larger than the
pCR®8/GW/TOPO® vector are assumed to contain insert.
59
bp
23,130
9416
6557
4361
2322
2027
564
23,130
9416
6557
4362
2322
2027
Figure 30. Colony screening via lysis and electrophoresis.
Eight colonies that possessed plasmids with insert of reasonable size, of at least
3500 bp, were further screened by colony PCR using GSP 2 along with either GW1 or
GW2 vector primer. By PCR screening the vectors from each colony using either
GW1 or GW2 in conjunction with GSP2, the directionality of the insert can be
discerned based on whether amplification occured with GW1 or GW2. Only four
fragments were amplified from plasmids of the eight colonies (Figure 31).
60
bp
A
C
B
2686
2000
1500
1200
1000
900
800
700
600
500
Figure 31. PCR screening of colonies with insert.
The isolated plasmids were then sequenced utilizing the appropriate vector
primer. Three of the four plasmids yielded 400-1023 bp of sequence. The first two
sequences were 90% identical to each other, but the third sequence was unrelated.
Homology searches agaist the NCBI nonredundant protein and nucleic acid databeases
for all sequences were performed with BLASTx and tBLASTx programs, respectively
(55). Two of three sequences were homologous to a secretory plant peroxidase and
the third had some homology to tyramine N-feruloyltransferase. The two peroxidase
sequences from colony A and B were very closely related exhibiting 90.7% sequence
identity (Table 6).
61
Table 6. Most homologous sequence based on BLASTx and tBLASTx searches of
NCBI protein and nucleotide databases of 3’ RACE products.
Colony
A
Sequence
length
1023
B
719
C
404
BLASTx
e value
tBLASTx
e value
properoxidase [Picea
abies]
properoxidase [Picea
abies]
PREDICTED: tyramine
N-feruloyltransferase
1e-78
Picea abies mRNA for
properoxidase
Picea abies mRNA for
properoxidase
Vitis vinifera tyramine
N-feruloyltransferase
4e-78
3e-85
3e-23
4e-78
2e-23
Because none of the sequences were highly homologous to monoterpene
synthase sequences, another round of 5’ RACE was performed, using the same 5’
RACE-ready cDNA template as before and using universal primer as the forward
primer and either degenerate primer, Block K or Block O, as reverse primers with the
same PCR program as before (Figure 32). These fresh PCR products were cloned
without gel purification as before. A total of 144 colonies were screened for inserts
based on electrophoretic analysis of vector size. From these colonies, 36 had an insert,
and these were subjected to PCR screening using vector primers to confirm insertion
of the amplification product. From the PCR screening, 20 of the 36 colonies, 10 from
Block K primer and 10 from Block O primer, possessed inserts of appropriate size
and were submitted for sequencing (Table 7). Again, none of these sequences were
obviously homologous to previously described monoterpene synthase sequences.
62
bp
1
2
3
2686
2000
1500
1200
1000
900
800
700
600
500
Figure 32. 5' RACE PCR products. Lane 1: 100 bp ladder (Fisher Scientific), Lane 2:
5’ RACE with Block K primer, Lane 3: 5’ RACE with Block O primer.
Table 7. Most homologous sequence based on BLASTx and tBLASTx searches of
NCBI protein and nucleotide databases of degenerate PCR products.
Colony
1
Sequence
length
940
2
BLASTx
e value
ADI61831.1
endonuclease-reverse
transcriptase [Bombyx mori]
9 e-12
937
No similarity found
n/a
3
222
No similarity found
n/a
4
855
hypothetical protein
STEHIDRAFT_161767
[Stereum hirsutum FP91666SS1]
8.3
5
962
XP_003520306.1
Predicted:
uncharacterized protein
LOC100807511 [Glycine max]
6
405
No similarity found
tBLASTx
e value
AC241283.1
Pinus taeda clone
PT_7Ba0038G01, complete
sequence
GU252851.1
Cercis canadensis clone 41
microsatellite sequence
Mus musculus chromosome 17
clone RP23-124G7, complete
sequence
emb|CR382122.1|
Kluyveromyces lactis strain
NRRL Y-1140 chromosome B
complete
4 e-24
8 e-11
JF330774.1
Capsicum annuum clone BAC
CaCM278G16, complete
sequence
1 e-13
n/a
AC185360.2
Populus trichocarpa clone
7 e-4
1.8
5 e-7
2.1
63
Pop1-53I10, complete
sequence
FP102047.7
Pig DNA sequence from clone
CH242-71K8 on chromosome
X
7
967
YP_710029.1
hypothetical protein
BAPKO_0614 [Borrelia afzelii
PKo]
2.4
0.85
8
948
GENE ID: 188283 T08D2.5 |
hypothetical protein
[Caenorhabditis elegans]
2.3
gb|AC124973.8| Mus
musculus strain C57BL/6J
clone rp23-329l13 map 19,
complete sequence
1.5
9
949
GENE ID: 6994889
CMU_027360 | hypothetical
protein [Cryptosporidium muris
RN66]
5.9
GENE ID: 100646622
LOC100646622 | solute carrier
family 35 member F5-like
[Bombus terrestris]
1.7
10
839
No similarity found
n/a
GI:389600251
Leishmania braziliensis
MHOM/BR/75/M2904
complete genome, chromosome
5
0.07
11
575
GENE ID: 11423145
MTR_1g006120 | Ribosomal
protein S10 [Medicago
truncatula]
1 e-38
dbj|AB029368.1|
Juniperus chinensis
mitochondrial gene for 18S
rRNA
2e-108
12
789
No similarities found
n/a
GENE ID: 459311
MPHOSPH10 | M-phase
phosphoprotein 10 (U3 small
nucleolar ribonucleoprotein)
[Pan troglodytes]
0.026
13
945
gb|ABK24819.1|
unknown [Picea sitchensis]
5e-8
gb|AC241346.1|
Pinus taeda clone
PT_7Ba4137F02, complete
sequence
1e-08
14
1030
BAB01972.1
copia-like retrotransposable
element [Arabidopsis thaliana]
2 e-8
AC241299.1
Pinus taeda clone
PT_7Ba2950E18, complete
sequence
7 e-37
15
871
ABK24819.1
unknown [Picea sitchensis]
4 e-8
AC241346.1
Pinus taeda clone
PT_7Ba4137F02, complete
sequence
1 e-08
64
16
734
XP_002515858.1
conserved hypothetical protein
[Ricinus communis]
>gb|EEF46527.1|
2 e-12
AC241348.1
Pinus taeda clone
PT_7Ba4180L06, complete
sequence
1 e-14
17
751
ZP_04455918.1
hypothetical protein
GCWU000342_0195[Shuttlew
orthia satelles DSM14600]
2.8
AM910996.2
Plasmodium
knowlesi strain
H chromosome 14,
complete genome
1.3
18
1112
XP_002515858.1
conserved hypothetical protein
[Ricinus communis]
>gb|EEF46527.1|
7 e-12
AC241348.1
Pinus taeda clone
PT_7Ba4180L06, complete
sequence
3 e-15
19
938
XP_002515858.1
conserved hypothetical protein
[Ricinus communis]
>gb|EEF46527.1|
4 e-12
AC241293.1
Pinus taeda clone
PT_7Ba2900E24, complete
sequence
3 e-15
20
947
ZP_03702608.1
NusA antitermination factor
[Flavobacteria bacterium
MS024-2A]
0.73
XM_001334017.3
PREDICTED: Danio rerio
NHS-like protein 2-like
(LOC794153)
0.22
To more exhaustively search for monoterpene synthase-related sequences, 3’
and 5’ RACE-Ready cDNA was resynthesized and 3’ RACE was performed with
degenerate forward primer Block C and 5’ RACE was performed with degenerate
primers Block K and Block O at an annealing temperature of 48 oC (Figure 33).
65
bp
4
1
2
3
2686
2000
1500
1200
1000
900
800
700
Figure 33. 3' and 5’ RACE PCR products. Lane 1: 100 bp ladder (Fisher-Scientific),
Lane 2: 3’ RACE with Block C primer, Lane 3: 5’ RACE with Block K primer, Lane
4: 5’ RACE with Block O primer.
Unlike the initial attempt, both the 5’ RACE and the 3’ RACE reactions yielded
products. Based on the position of the conserved domains within known monoterpene
synthase genes, both 3’ RACE with Block C and 5’ RACE with Block O yielded
fragments near the expected size of ~ 1700bp; however, 5’ RACE with Block K did
not yield a fragment of the expected 1200 bp size.
To increase the probability of cloning a RACE product of the size expected for
a monoterpene synthase fragment, bands of the expected size were isolated and
purified from a low-melt agarose gel before cloning. Gel-purified fragments were
then cloned as before. This time there were fewer colonies present, which was
66
expected, as the PCR product was more dilute due to the presence of agarose from the
purification step with low melt agarose.
Nineteen colonies from the 3’ RACE with Block C primer and 20 colonies
from the 5’ RACE with Block O primer were screened for the presence of inserts
based on plasmid size. Twelve of the 3’ RACE colonies had an insert present, and 5
of the 5’ RACE with Block O primer had an insert present. These colonies were then
subjected to PCR screening, which showed that only five 3’ RACE colonies had
inserts of the appropriate size. Isolated plasmids were sent for sequencing, and four of
the five colonies afforded sequence (Table 8).
Table 8. Sequence analysis from gel purified 3’ RACE PCR.
Colony
1
Sequence
length
937
BLASTx
e value
tBLASTx
XP_003520302.1
PREDICTED: pyruvate
kinase, cytosolic isozymelike [Glycine max]
2e-5
FJ603644.1
Taxus cuspidata 3'-Ndebenzoyltaxol Nbenzoyltransferase
e
value
2e-65
2
942
BAB01972.1
copia-like retrotransposable
element [Arabidopsis
thaliana]
2 e-8
AC241299.1
Pinus taeda clone
PT_7Ba2950E18, complete
sequence
7 e-37
3
936
No similarity found
n/a
FR796467.1
Leishmania infantum JPCM5
genome chromosome 35
0.63
4
345
No similarity found
n/a
dbj|AK396764.1| Sus scrofa
mRNA, clone: PST010087B10,
expressed in prostate
0.41
67
In summary, a total of 39 amplification products were sequenced from all the
RACE reactions, of which 27 had some homology to proteins on the NCBI protein
database. However, none had obvious homology to previously described monoterpene
synthase.
Many studies with conifers have utilized homology-based PCR in order to
successfully isolate terpene synthase genes (21, 35, 49-53). Our study, like those of
other authors, involved isolating mRNA and designing dengenerate primers using
conserved domains in homologous sequences. However, most of the previous studies
constructed lamda cDNA libraries that were probed with sequences amplified with
dengererate primers and we did not. Because a cDNA library was not constructed the
sampling of colonies was random, wherereas a cDNA library allows for the use of the
degenerate primers to probe for potential candidates. The absence of a cDNA library
makes finding the gene of interest more difficult and less probable, even though we
did try to maximize the probability of isolating a monoterpene synthase gene by gel
purification of PCR products.
In the other studies, RNA was isolated from bark, shoot tissue or sapling stems
(21, 35, 49-53), whereas we used callus cultures. Callus cultures presented challenges,
not only in mRNA isolation due to their high polysaccharide and polyphenol content,
but also in their initiation and maintenance. Calli are prone to mold, especially during
initiation, but also when replating. Each time the calli needed to be replated (every 46 weeks), some succumbed to mold development. Due to mold issues, at the RNA
68
isolation stage, there were not enough calli to perform multiple RNA isolation
attempts, which might have afforded higher quality mRNA.
3.7 Conclusions
A homology based PCR strategy was used to try to isolate a monoterpene
synthase gene involved in the biosynthesis of -thujaplicin from C. dupreziana callus
culture. Although an obvious monoterpene synthase gene candidate was not
identified, several significant goals were accomplished.
We developed a robust system for studying -thujaplicin biosynthesis. This is
the first report of C. dupreziana callus cultures being used for -thujaplicin
investigation; most investigations of -thujaplcin with callus cultures use C. lusitanica
calli. These C. dupreziana callus cultures were initiated and have been maintained for
over three years and have been shown to actively produce -thujaplicin whereas
unelicited callus cultures of C. lusitanica have been reported to produce no thujaplicin (47).
Additionally, a method for GC-MS analysis of -thujaplicin that is less
sensitive to column conditions was developed. By derivatization with BSFTA+1%
TMSC, -thujaplicin can be analyzed even with a suboptimum column. This is the
first report of analysis of derivatized -thujaplicin.
Finally, we also developed a method for RNA isolation from callus cultures.
Because of the high polysaccharide and polyphenol levels present in callus cultures, it
69
was necessary to incorporate a 10% ethanol precipitation to remove polysaccharides,
EDTA and thiourea to inhibit polyphenoloxidases and aurintricarboxylic acid to
inhibit RNases.
In spite of these accomplishments, a cDNA with high homology to a
monoterpene synthase was not isolated. From the GC-MS analysis of the
monoterpene content we know that monoterpene synthases must be present, even if we
were unable to isolate a monoterpene synthase sequence. The failure to isolate an
apparent monoterpene synthase cDNA may be due to several factors. First, the
degenerate primers we used may not have the appropriate sequence for amplifying a
monoterpene synthase cDNA from Cupressus species. In this study we used three
degenerate primers, one forward primer (Block C) and two reverse primers (Block K
and Block O). The first successful isolation of a monoterpene synthase sequence from
a gymnosperm via similarity based PCR strategy used four primers, three forward and
one reverse, and cDNA from a phage library as the template (35). Only one primer
was successful. Another study utilized 22 primers to attempt to amplifiy product (49).
For C. dupreziana, designing a bigger pool of degenerate primers or even using
primers from other studies isolating monoterpene synthase sequence that have proven
successful may result in amplification of our sequence of interest. Additionally, the
other studies employed a “broad range of amplification conditions” (35), and more
optimization of annealing temperatures with our primers may lead to amplification of
a monoterpene synthase sequence. Alternatively, monoterpene synthases in
70
Cupressaceae may be substantially different from other monoterpene synthase
sequences, so that some of the sequences obtained actually encode a monoterpene
synthase.
Amplification may also not have been successful due to a low abundance of
monoterpene synthase in mRNA preparations. Isolating mRNA from methyl
jasmonate-elicited cultures may increase the abundance of monoterpene synthase
transcripts in the mRNA template, increasing the probability of successful
amplification of monoterpene cDNA. Further optimization of the RNA isolation
protocol may also lead to a higher quality RNA template.
Finally, although less probable, amplification of an apparent monoterpene
synthase cDNA may not have been successful because monoterpene synthases from
Cupressaceae may be so divergent from the other conifer monoterpene synthases that
a homology-based approach is less likely to succeed. Using a more diverse collection
of sequences that include non-conifer monoterpene synthases in the CODEHOP
primer design process would broaden the potential range of monoterpene synthase
sequences potentially amplified by the designed degenerate primers.
71
REFERENCES
1.
Anderson, A. B., Scheffer, T., and Duncan, C. G. (1963) The chemistry of
decay resistance and its decrease with heartwood aging in incense cedar
(Libocedrus decurrens Torrey), Holzforschung-International Journal of the
Biology, Chemistry, Physics and Technology of Wood 17, 1-5.
2.
Barton, G., and MacDonald, B. (1971) The Chemistry and Utilization of
Western Red Cedar, Publication.
3.
Yamaguchi, T., Fujita, K., and Sakai, K. (1999) Biological activity of extracts
from Cupressus lusitanica cell culture, Journal of Wood Science 45, 170-173.
4.
Aharoni, Y., Copel, A., and Fallik, E. (1993) Hinokitiol (ß-thujaplicin), for
postharvest decay control on Galia melons, New Zealand Journal of Crop and
Horticultural Science 21, 165-169.
5.
Arima, Y., Hatanaka, A., Tsukihara, S., Fujimoto, K., Fukuda, K., and Sakurai,
H. (1997) Scavenging activities -, -and-thujaplicins against active oxygen
species, Chemical and Pharmaceutical Bulletin 45, 1881-1886.
6.
MATSUMURA, E., MORITA, Y., DATE, T., TSUJIBO, H., YASUDA, M.,
OKABE, T., ISHIDA, N., and INAMORI, Y. (2001) Cytotoxicity of the
hinokitiol-related compounds, -thujaplicin and -dolabrin, Biological &
Pharmaceutical Bulletin 24, 299-302.
7.
Trust, T., and Coombs, R. (1973) Antibacterial activity of β-thujaplicin,
Canadian Journal of Microbiology 19, 1341-1346.
8.
Rennerfelt, E. (1948) Investigations of Thujaplicin, a Fungicidal Substance in
the Heartwood of Thuja plicata D. Don, Physiologia Plantarum 1, 245-254.
9.
Sowder, A. (1929) Toxicity of water-soluble extractives and relative durability
of water-treated wood flour of western red cedar, Industrial & Engineering
Chemistry 21, 981-984.
10.
Cartwright, K. (1941) The Variability in Resistance to Decay of the Heartwood
of Home-grown Western Red Cedar (Thuja plicata d. don) and its Relation to
Position in the Log., Forestry 15, 65.
72
11.
Anderson, A. B. (1955) Recovery and Utilization of Tree Extractives,
Economic Botany 9, 108-140.
12.
Croteau, R., Kutchan, T. M., and Lewis, N. G. (2000) Natural products
(secondary metabolites), Biochemistry and Molecular Biology of Plants, 12501318.
13.
Zhao, J. (2007) Plant troponoids: chemistry, biological activity, and
biosynthesis, Current Medicinal Chemistry 14, 2597-2621.
14.
Pauson, P. L. (1955) Tropones and tropolones, Chemical Reviews 55, 9-136.
15.
Dewar, M. (1945) Structure of stipitatic acid, Nature 155, 50-51.
16.
Kurihara, T., Mine, H., Satoh, Y., Wakabayashi, H., Motohashi, N., and
Sakagami, H. (2006) Relationship between electronic structure and cytotoxic
activity of tropolones, In Vivo 20, 391-395.
17.
Bentley, R. (2008) A fresh look at natural tropolonoids, Natural Product
Reports 25, 118-138.
18.
Lee, M. J., Kim, J. W., and Yang, E. G. (2010) Hinokitiol activates the
hypoxia-inducible factor (HIF) pathway through inhibition of HIF
hydroxylases, Biochemical and Biophysical Research Communications 396,
370-375.
19.
Arima, Y., Nakai, Y., Hayakawa, R., and Nishino, T. (2003) Antibacterial
effect of β-thujaplicin on staphylococci isolated from atopic dermatitis:
relationship between changes in the number of viable bacterial cells and
clinical improvement in an eczematous lesion of atopic dermatitis, Journal of
Antimicrobial Chemotherapy 51, 113.
20.
Zavarin, E., Smith, L. V., and Bicho, J. G. (1967) Tropolones of
Cupressaceae--III, Phytochemistry 6, 1387-1394.
21.
Bohlmann, J., Phillips, M., Ramachandiran, V., Katoh, S., and Croteau, R.
(1999) cDNA cloning, characterization, and functional expression of four new
monoterpene synthase members of the Tpsd gene family from grand fir (Abies
grandis), Archives of Biochemistry and Biophysics 368, 232-243.
73
22.
Cheng, A. X., Lou, Y. G., Mao, Y. B., Lu, S., Wang, L. J., and Chen, X. Y.
(2007) Plant terpenoids: biosynthesis and ecological functions, Journal of
Integrative Plant Biology 49, 179-186.
23.
Bohlmann, J., Meyer-Gauen, G., and Croteau, R. (1998) Plant terpenoid
synthases: molecular biology and phylogenetic analysis, Proceedings of the
National Academy of Sciences 95, 4126.
24.
Ono, M., Asai, T., and Watanabe, H. (1998) Hinokitiol production in a
suspension culture of Calocedrus formosana Florin, Bioscience, Biotechnology,
and Biochemistry 62, 1653-1659.
25.
Ramachandra Rao, S., and Ravishankar, G. (2002) Plant cell cultures:
Chemical factories of secondary metabolites, Biotechnology Advances 20, 101153.
26.
Sakai, K., Kusaba, K., Tsutsumi, Y., and Shiraishi, T. (1994) Secondary
metabolites in cell culture of woody plants, 3: Formation of -thujaplicin in
Cupressus lusitanica callus cultures treated with fungal elicitors, Journal of the
Japan Wood Research Society 40.
27.
Inada, S., Tsutsumi, Y., and Sakai, K. (1993) Elicitor of the -thujaplicin
accumulation in callus cultures of Cupressus lusitanica, Journal of the Faculty
of Agriculture-Kyushu University 38.
28.
Zhao, J., Zheng, S. H., Fujita, K., and Sakai, K. (2004) Jasmonate and ethylene
signalling and their interaction are integral parts of the elicitor signalling
pathway leading to -thujaplicin biosynthesis in Cupressus lusitanica cell
cultures, Journal of Experimental Botany 55, 1003-1012.
29.
Yamada, J., Fujita, K., and Sakai, K. (2002) Feedback regulation of thujaplicin production and formation of its methyl ether in a suspension culture
of Cupressus lusitanica, Phytochemistry 60, 447-450.
30.
Yamaguchi, T., Itose, R., and Sakai, K. (1997) Biosynthesis of heartwood
tropolones, 1: Incorporation of mevalonate and acetate into -thujaplicin
(hinokitiol) of Cupressus lusitanica cell cultures, Journal of the Faculty of
Agriculture-Kyushu University 42.
31.
Fujita, K., Yamaguchi, T., Itose, R., and Sakai, K. (2000) Biosynthetic
Pathway of -Thujaplicin in the Cupressus lusitanica Cell Culture, Journal of
Plant Physiology 156, 462-467.
74
32.
Zhao, J., Fujita, K., and Sakai, K. (2001) Biochemistry & Molecular BiologyProduction of -Thujaplicin in Cupressus lusitanica Suspension Cultures Fed
with Organic Acids and Monoterpenes, Biosciences Biotechnology and
Biochemistry 65, 1027-1032.
33.
Matsunaga, Y., Fujita, K., Yamada, J., Ashitani, T., and Sakai, K. (2003)
Monoterpenes produced by Cupressus lusitanica cultured cells including a
novel monoterpene (1 S, 2 S, 6 S)-(+)-1, 6-epoxy-4 (8)-p-menthen-2-ol,
Natural Product Research 17, 441-443.
34.
Zhao, J., Matsunaga, Y., Fujita, K., and Sakai, K. (2006) Signal transduction
and metabolic flux of -thujaplicin and monoterpene biosynthesis in elicited
Cupressus lusitanica cell cultures, Metabolic Engineering 8, 14-29.
35.
Bohlmann, J., Steele, C. L., and Croteau, R. (1997) Monoterpene synthases
from grand fir (Abies grandis), Journal of Biological Chemistry 272, 21784.
36.
Murashige, T., and Skoog, F. (1962) A revised medium for rapid growth and
bio assays with tobacco tissue cultures, Physiologia Plantarum 15, 473-497.
37.
Kistner, C., and Matamoros, M. (2005) RNA isolation using phase extraction
and LiCl precipitation, Lotus japonicus Handbook, 123-124.
38.
Simms, D., Cizdziel, P. E., and Chomczynski, P. (1993) TRIzol: A new
reagent for optimal single-step isolation of RNA, Focus 15, 532-535.
39.
Lewinsohn, E., Steele, C. L., and Croteau, R. (1994) Simple isolation of
functional RNA from woody stems of gymnosperms, Plant Molecular Biology
Reporter 12, 20-25.
40.
Sambrook, J., and Russell, D. W. (2001) Molecular Cloning: a laboratory
manual, Vol. 2, CSHL press.
41.
Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T. J., Higgins, D. G.,
and Thompson, J. D. (2003) Multiple sequence alignment with the Clustal
series of programs, Nucleic Acids Research 31, 3497-3500.
42.
Rose, T. M., Henikoff, J. G., and Henikoff, S. (2003) CODEHOP (COnsensusDEgenerate hybrid oligonucleotide primer) PCR primer design, Nucleic Acids
Research 31, 3763-3766.
75
43.
Chenchik, A., Zhu, Y. Y., Diatchenko, L., Li, R., Hill, J., and Siebert, P. D.
(1998) Generation and use of high-quality cDNA from small amounts of total
RNA by SMART PCR, Gene Cloning and Analysis by RT-PCR, 305-319.
44.
Barnes, W. M. (1977) Plasmid detection and sizing in single colony lysates,
Science 195, 393-394.
45.
Saris, P. E. J., Paulin, L. G., and Uhlen, M. (1990) Direct amplication of DNA
from colonies of Bacillus subtilis and Escherichia coli by the polymerase chain
reaction, Journal of Microbiological Methods 11, 121-126.
46.
Zhao, J., Fujita, K., Yamada, J., and Sakai, K. (2001) Improved ß-thujaplicin
production in Cupressus lusitanica suspension cultures by fungal elicitor and
methyl jasmonate, Applied Microbiology and Biotechnology 55, 301-305.
47.
De Alwis, R., Fujita, K., Ashitani, T., and Kuroda, K. (2009) Induced
monoterpene and lignin production in mechanically stressed and fungal elicited
cultured Cupressus lusitanica cells, Plant Biotechnology Reports 3, 57-65.
48.
Schneiderbauer, A., Sandermann, H., and Ernst, D. (1991) Isolation of
functional RNA from plant tissues rich in phenolic compounds, Analytical
Biochemistry 197, 91-95.
49.
Huber, D. P. W., Philippe, R. N., Godard, K. A., Sturrock, R. N., and
Bohlmann, J. (2005) Characterization of four terpene synthase cDNAs from
methyl jasmonate-induced Douglas-fir, Pseudotsuga menziesii, Phytochemistry
66, 1427-1439.
50.
Schmidt, A., and Gershenzon, J. (2008) Cloning and characterization of two
different types of geranyl diphosphate synthases from Norway spruce Picea
abies, Phytochemistry 69, 49-57.
51.
Jones, C. G., Keeling, C. I., Ghisalberti, E. L., Barbour, E. L., Plummer, J. A.,
and Bohlmann, J. (2008) Isolation of cDNAs and functional characterisation of
two multi-product terpene synthase enzymes from sandalwood, Santalum
album L, Archives of Biochemistry and Biophysics 477, 121-130.
52.
Faldt, J., Martin, D., Miller, B., Rawat, S., and Bohlmann, J. (2003) Traumatic
resin defense in Norway spruce (Picea abies): methyl jasmonate-induced
terpene synthase gene expression, and cDNA cloning and functional
characterization of (+)-3-carene synthase, Plant Molecular Biology 51, 119133.
76
53.
Vogel, B. S., Wildung, M. R., Vogel, G., and Croteau, R. (1996) Abietadiene
synthase from grand fir (Abies grandis) cDNA isolation, characterization, and
bacterial expression of a bifunctional diterpene cyclase involved in resin acid
biosynthesis, Journal of Biological Chemistry 271, 23262-23268.
54.
Davis, E., and Croteau, R. (2000) Cyclization enzymes in the biosynthesis of
monoterpenes, sesquiterpenes, and diterpenes, Biosynthesis, 53-95.
55.
Altschul, S. F., Madden, T. L., Scheffer, A. A., Zhang, J., Zhang, Z., Miller,
W., and Lipman, D. J. (1997) Gapped BLAST and PSI-BLAST: a new
generation of protein database search programs, Nucleic Acids Research 25,
3389-3402.