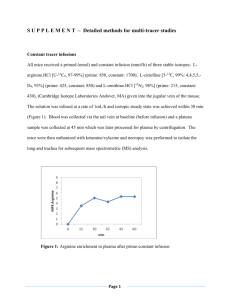
AA Disorder table
advertisement

Disease Cystinuria Type A Mutation Cationic AA & Cystine transporter Mutation in heavy chain Mutation in light chain Type B Type AB Mutations in both chains Hartnup Disorder Neutral AA transporter (poor tryptophan uptake) Kidney Form Transporter does not associate with collectrin— not expressed on apical surface of kidneys –AAs not reabsorbed Small Intestine Form ACE 2 necessary for expression of transporter in SI Symptoms -excrete large amounts of cystine in urine -flat hexagonal crystals in urine - + in cyanin-nitroprusside test (purple) - cystine not reabsorbed in kidney (kidney stones) -excrete large amounts of cystine, lysine, arginine, and ornithine -homoz-excrete large amounts of cystine, lysine, arginine, and ornithine -heteroz-slightly affected -occasionally get cystine kidney stones -compound heterozygote--full blown cystinuria Similar to pellagra -rash w/ photosensitivity -psychosis -cerebellar ataxia -elevated AAs in urine -Faulty tryptophan uptake **precursor to nicotinamide (rash/photosensitivity) **precursor to serotonin (psychosis) -many patients are asymptomatic **protein deficient diet can make it worse Treatment -adequate hydration-keeps urine dilute -keep urine slightly alkaline-↑cystine solubility **oral Kcitrate, Na citrate, & Na Bicarb -reduction of cysteine & methinonin—reduce animal protein -drugs—penicilliamine and αmercaptopropionylglycine **reacts w/ cysteine and make a more soluble disulfide bond -dietary niacin *replaces deficient nicotinamide Lysinurgic Protein Intolerance Maple Syrup Urine Disease LAT 1 Branched Chain Amino Acid DHD E1-ketoacid decarboxylase (type 1A or 1B) (thiamine dependent) E2-dihydrolipoyl acyltransferase (type II) -defective uptake of lysine, arginine, and ornithine in small intestines and kidneys -↑ urinary excretion of lysine, arginine, and ornithine -↓ levels of plasma lysine and arginine - derangement in urea cycle in liver (due to low ornithine) *elevation of plasma ammonia *neurological problems *aversion to protein rich foods—vomiting/diarrhea -growth retardation, enlargement of spleen, muscular hypotonia, and osteoporosis -renal and pulmonary complications **infants symptom free as long as breast fed** -enchephalopathy 4-7 days after birth -branched chain AAs elevated in blood **valine, isoleucine, and leucine** -alloleucine present (diagnostic) -lethargy/no interest in feeding -weight loss -progressive neurological deterioration -alternate hypo- and hypertonia -burnt sugar or maple odor in urine -some degree of mental impairment E3-dihydrolipoaminde dehydrogenase (t 3) Isovaleric Acidemia Isovaleryl CoA Dehydrogenase (IVA) (coenzyme: Flavin) catalyzes isovaleryl Coa β-methylcotonoyl CoA -accumulation of isovaleric acid and 3-hydroxyvaleric acid in plasma and urine First type -symptoms seen w/in 2 wks of birth -vomiting, dehydration, listlessness, acidosis Second Type -chronic and intermittent -restrict protein intake (make sure px still receive essential AAs) -promote N excretion (Na benzoate and phenylbuturate) -oral citrulline (precursor to ornithine--up take through other neutral AA transporter) -must remove toxic metabolites -continuous blood exchange transfusion or dialysis -thiamine may improve tolerance in some patients (type 1A and 1B) -pxs generally healthy between episodes of metabolic imbalance -degree of impairment depends on amount of time leucine levels were high -dietary protein restriction—limit intake of leucine, but supplement with mixture of AAs -oral glycine and IV carnitine during metabolic crisis: reacts w/ IA to form IA-carnitine and IA-glycine ***non toxic*** -early diagnosis and treatment Isovaleric Acidemia (continued) Methylmalonic Acidemia (MMA) & Propionic Acidemia (PA) Methylmalonyl CoA mutase -catalyzes methylmalonyl CoA Succinyl CoA -adenosylcobalmin is cofactor Propionyl-CoA carboxylase -catalyzes propionyl CoA methylmalonyl CoA -Biotin as cofactor Nonketonic glycine cleavage system hyperglycinemia -vomiting, lethargy (progressing to coma) -elevated anion gap -metabolic acidosis -ketonuria -unwashed body odor -exacerbated in first 12mos by stress, infection, and high protein meals -gastrointestinal signs; poor suckling, vomiting, weight loss, abdominal distention -neurological manifestations; abnormal posture & movement, generalized hypotonia, lethargy, seizures -metabolic acidosis -increased anion gap (due to increased organic acids) -ketonuria -anemia -hyperuricemia (↑ uric acid) -leukopenia -hyperammoniemia -brain: accumulation of metabolites inhibit TCA and mitochondrial respiration; necrosis; spongioisis, cyst formation -kidney: in MMA—chronic renal failure -heart: in PA—cardiomyopathy (rapidly fatal); appears to be related to poor uptake of carnitine -↑ in glycine -glycine receptors in inhibitory neurons (skeletal musc) -NMDA receptor has glycine binding site (↑ Ca influx) -decreased supply of N5N10CH2THF -derangement of 1 C metabolism interferes w/ remethylation of methionine -lethargy & vomiting -convulsions & loss of primitive reflexes -myoclonic seizures -apnea minimizes effects on development of brain **better long term diagnosis than any other organic aciduria** -outlook is very bad -limit dietary protein -avoid long fasts -kidney/liver transplant -reduce activity of NMDA glutamate receptor (diazepam: competes with glycine for allosteric site; ketamine and dextropmethorphan: NMDA receptor antagonists -promote excretion of glycine w/ Na benzoate (forms Hippurate) -control seizures (phenobarbital) Urea Cycle Disorder Hyperammonemia type 1 Hyperammonemia Type 2 Reaction affected NH3+HCO3- carbamoyl PO4 Ornithinecitrulline Enzyme deficient Carbamoyl Phosphate Synthetase I (CPI) Treatment Arginine (stimulates CPI) N-Acetylglutamate synthase Carbamoy glutamate (activates CPI) ↑carb/↓protein diet Ornithine transcarbamylase Benzoic acid or phenylacetic acid to treat ammonia intoxication Arginine (enhances citrulline excretion) Arginosuccinate synthetase deficiency Citrulline+aspartate arginosuccinate Arginosuccinate synthetase Arginosuccinate lyase deficiency Arginase Deficiency Arginosuccinate arginine + fumarate Arginine orthinine + urea Arginosuccinate lyase arginine Arginase Diet of essential AAs (no arginine) Low protein diet to reduce plasma ammonium Treatments for all of the urea cycle diseases include: -low protein/high carb diet: reduces ammonia production -levulose: bacteria in gut metabolize levulose to acidic compoundmore ammonium—excreted in feces -antibiotics: suppress ammonia producing bacteria in gut -Sodium Benzoate/sodium phenylbutyrate: form covalent products with glycine (hippurate) and glutamine(phenylacetyl glutamate)— promote nitrogen excretion if feces