ISOLATION OF CYTOPLASMIC AND OUTER MEMBRANE
advertisement

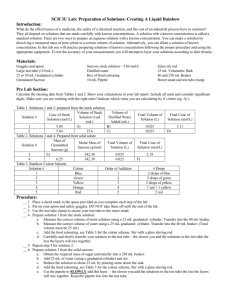
ISOLATION OF CYTOPLASMIC AND OUTER MEMBRANE FRACTIONS (Method by Osborn et al., 1972, J. Biol. Chem. 247: 3962-3972) What is needed: Reserve the shaker. Need 1L baffled flasks with media autoclaved in them. Rinse the flasks 6 times with distil water before use. Examine for dirt. Need 6 each of 500ml centrifuge bottles. Need ultra centrifugation tubes for T45i , SW31 and T75i rotors Rotors are kept in K155 cold room. Inspect their o rings. Locate where lysozyme, DNAse and RNAse are kept. Make all the buffers (note that the pH of the buffer can ranges from 7-8. It does not matter at the end). 0.5M sucrose, 20mM Tris-HCl, pH 8 0.25M sucrose, 20mM Tris-HCl, pH 8 (make this by diluting the previous buffer by 2X) Ice cold water. 10mM Tris, pH 8 25mg/mL lysozyme. 10mM Na-EDTA, pH 8. 1M MgCl2, 10X PIC (protease inhibitor tablets) 100uL 2mg/mL DNase 20uL 10mg/mL RNase 20% sucrose (w/w), 1mM EDTA, pH 8, 10mM Tris, pH 8 73% sucrose, 1mM EDTA, pH 8, 10mM Tris, pH 8 53% sucrose 1mM EDTA, pH 8, 10mM Tris, pH 8 20% sucrose 1mM EDTA, pH 8, 10mM Tris, pH 8 How to inoculate bacteria: consider OD 1= 5X10^8 for both PA and Acinetobacter and 10^ for Salmonella or E. coli. Inoculate at 0.02. With 4 doublings, you should get into around 0.3 OD. For polymyxin or C18G treatment, add the compound at OD= 0.2. For CAMP experiments, harvest at 6090 min post exposure. Media, LB, minimal or BHI: To prevent autolysis, add MgCl2 to 1mM for growing Acinetobacter. 1. Inoculate 10mL O/N culture or a plate with a lawn of bacteria. 2. Inoculate 250ml to 1L with resuspended bacteria from the plate or from the ON culture- grow at 37°C until it is grown enough for the experiment. 3. Centrifuge cells at 8000g, 8 min, 4°C in the Sorvall (room K113) or Beckman (room K110) using a 500ml Nalgene bottle. Pour off media and invert bottles onto a paper towel to remove residual culture medium. 4. Resuspend cell pellet rapidly in 25mL ice cold hypertonic solution (0.5M sucrose, 20mM Tris-HCl, pH 8). Make sure it is well suspended! Plasmolysis occurs at this stage. Add a small stir bar. 5. Add 150μL of freshly prepared 25mg/mL lyzozome (final concentration: 150μg/mL) and incubate on ice for 5min. 6. Form spheroplasts by diluting suspension with 20ml cold 10mM Na-EDTA, pH 8 and stir for 10min (spheroplasts are usually formed in <10min when the lysozyme destroys the cell wall). Bacteria are susceptible to lysis if the medium become hypotonic at this stage. NOTE: FOR PSEUDOMONAS, DON’T ADD EDTA. Instead, add 25ml ice cold water. 7. Stabilize spheroplasts by adding 5mL 1M MgCl2 (final concentration: ~20mM Mg2+). Mix by swirling. Bacteria are now resistant to lysis. 8. Centrifuge spheroplasts at 8000g, 10min, 4°C (in a benchtop centrifuge). Remove the supernatant out and drain well. 9. Resuspend spheroplasts in 25 mL (per liter initial culture) of 0.25M sucrose, 10mM Tris. 10. Add the following to the resuspended spheroplasts: Crush one tablet of PIC in 1ml buffer, dissolve, add to the sphepst. 100uL 2mg/mL DNase 20uL 10mg/mL RNase MgCl2 to 1mM (25ul of 1M MgCl2) 11. Lyse the spheroplasts by running them 3X in Avastin homogenizer (Xena). 12. Add 250ul 0.5M EDTA to the lysate. Centrifuge lysates at 8,000g, 10min, 4°C in Beckman Allegra in K126 or Napco in K116/K155 to remove intact cells. Save the supernatant into a new conical. Discard the pellet. 13. Centrifuge the supernatant fraction at 38K, 1hr, 4°C in Ti 45 rotor in the ultracentrifuge. (STOP POINT: Alternatively you may spin overnight at 35K) 14. Resuspend the membrane pellet in 4mL 20% sucrose (w/w), 1mM EDTA, pH 8, 10mM Tris, pH 8, with Teflon sticks. Homogenize using Teflon plunger. 15. Set up sucrose gradient in ultra-clear SW41 ultra-centrifuge tubes by adding solutions carefully and slowly in the following order (all solutions at 4°C): (see the method of making the gradient at the end of this article). 2mL 73% sucrose 4mL 53% sucrose ~2mL total membrane in 20% sucrose (or ½ total membrane) ~4mL 20% sucrose (or to top of tube) 16. Centrifuge at 37K, ≥12hrs. 4°C in the SW41 rotor in the ultra-centrifuge. Longer centrifugation time results in better separation. 17. Carefully collect the inner membrane fractions with a Pasteur pipette. 18. Pierce the bottom of the tube with a needle, let the buffer drain by drop wise (control the flow by using the thumb to close the top of the tube). When the outermembrane ring is at the hole, let it drop into a 15ml falcon tube. ***Membranes can be stored at this point*** REMOVAL OF SUCROSE AND DETERMINING YIELD OF MEMBRANE FRACTIONS 19. Add approximately 1mL membranes to 6mL 10mM Tris, pH 8, 1mM EDTA buffer. Mix with a Teflon stick. 20. Centrifuge at 50K, 1hr, 4°C in the Ti70 rotor in the ultra-centrifuge. Decant the supernatant. 21. Resuspend membranes in ~700μL 10mM Tris, 1mM EDTA buffer and mix with Teflon sticks. Homogenize with Teflon plunger. 22. Store membranes on ice and use the PIERCE Coomassie Protein Assay to determine the protein concentration of the membrane fractions (1μL, 2μL, and 5μL samples). 23. Aliquot the membranes and quick-freeze them in a dry ice/EtOH bath. Store the membranes at -80°C. ***ALL SOLUTIONS SHOULD BE STERILE AND AT 4°C*** Sucrose density gradient. The gradient is prepared by layering progressively less dense sucrose solutions upon one another; therefore the first solution applied is the 73 % sucrose solution. A steady application of the solutions yields the most reproducible gradient. Firstly the Beckman tube is held upright in a tube stand. Next a 200 μl pipettor tip is placed on the end of a 1000 μl pipettor tip. Both snugly fitting tips are held steady by a clamp stand and the end of the yellow tip is allowed to make contact with the inside wall of the tube as shown below. Now sucrose solutions can be placed inside the blue tip and gravity will feed the solutions into the tube slowly and steadily, starting with the 73% solution. if the solutions fail to feed down through the tips and into the tube a small amount of positive air pressure can be applied to the blue tip to start the flow. This is done by gently tapping on the wide end of the blue tip. Once the 73 % solution has drained into the tube, the 53 % solution can be loaded into the blue tip which will then flow down the inside of the tube and layer on top of the 73 % solution. References: (i) LYSIS OF GRAM-NEGATIVE ORGANISMS AND THE ROLE OF VERSENE* ROY REPASKE. Department of Bacteriology, Indiana University, Bloomington, Ind. (U.S.A,) VOL. 30 (1958) BIOCHIIMICA ET BIOPHYSICA ACT. Page 225 (2) HOW DOES LYSOZYME PENETRATE THROUGH THE I~ACTERIAL OUTEK MEMBRANE? BERNARD W]THOLT, HARM VAN HEERIKRUIZEN and LOE DE LEIJ. Biochimica el Biophysica Acta, 443 (1976) 34-44 (3) Osmotic Pressure in Escherichia coli as Rendered Detectable by Lysozyme Attack. PAUL SCHEIE. J of Bacteriology, May 1973, p. 549-555; Vol. 114, No. 2 (4) Mechanism of Assembly of the Outermembrane of Salmonella typhimuriu: Isolation and characterization of cytoplasmic and outer membrane. M. J. Osborn, J. E. Gander, E. Parisi JBC 1972, 247,:3962