Additional Methods
advertisement

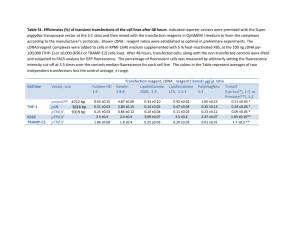
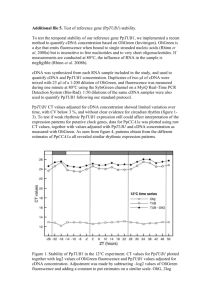
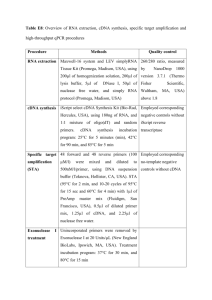
ADDITIONAL METHODS All protein fusions To construct the mEos4a fusions to the C-terminus of the fluorescent protein (number of linker amino acids in parenthesis), the subsequent digests were performed: human lamin A (18), NheI and BglII (cDNA source: David Gilbert, FSU; NM_170707.2); human β-actin (18), NheI and BglII (cDNA source: Clontech, Mountain View, CA; NM_001101.3); human light chain clathrin (15), NheI and BglII (cDNA source: George Patterson, NIH; NM_001834.2); human α-tubulin (18), NheI and BglII (cDNA source: Clontech; NM_006082); human H2B (10), BglII and NheI (histones, cDNA source: George Patterson, NIH; NM_021058.3). To prepare the Nterminal fusions to mEos4a, the following digests were performed: human keratin (18), EcoRI and NotI (cDNA source: Open Biosystems, Huntsville, AL; NM_199187.1); human calnexin (14), AgeI and NotI (cDNA source: Origene, Rockville, MD; NM_001746.3); human zyxin (6), BamHI and NotI (cDNA source: Origene; NM_003461); human caveolin 1 (10), NheI and BglII (cDNA source: Origene; NM_001753); human H2B (6), BamHI and NotI (histones, cDNA source: George Patterson, NIH; NM_021058.3). To construct the mEos4b fusions to the C-terminus of the fluorescent protein (number of linker amino acids in parenthesis), the following digests were performed: human H2B (6), BglII and NheI (histones, cDNA source: George Patterson, NIH; NM_021058.3); human β-actin (18), NheI and BglII (cDNA source: Clontech; NM_001101.3); human myotilin (14), AgeI and BspEI (cDNA source: Origene; NM_006790.1); mouse CAF1 (10), AgeI and BspEI (chromatin assembly factor, cDNA source: Akash Gunjan, Florida State University; NM_013733.3); human α-tubulin (18), NheI and BglII (cDNA source: Clontech; NM_006082); human lysosomal membrane glycoprotein 1 (20), BamHI and NotI (LAMP1, George Patterson, NIH; NM_012857.1); human lamin A (18), NheI and BglII (cDNA source; David Gilbert, FSU; NM_170707.2); mouse myosin IIA (14), NheI and BglII (cDNA source: Origene; NM_022410.2); human light chain clathrin (15), NheI and BglII (cDNA source: George Patterson, NIH; NM_001834.2); human zyxin (6), BamHI and NotI (cDNA source: Origene; NM_003461); human TOMM20 (10), BamHI and NotI (translocase outer mitochondria membrane 20, cDNA source: Origene; NM_014765.2); human caveolin 1 (10), NheI and BglII (cDNA source: Origene; NM_001753); human CENP-B (22), BamHI and NotI (cDNA source: Alexey Khodjakov, Wadsworth Center, NY; NM_001810.5); rat connexin-43 (7), BamHI and NotI (cDNA source: Matthias Falk, Lehigh U; NM_001004099.1); chicken c-src (7), BamHI and EcoRI (cDNA source: Marilyn Resh, Sloan-Kettering Institute, New York; XM_001232484.1); CytERM (17), AgeI and NotI (rabbit cytochrome p450, Erik Snapp, Albert Einstein College of Medicine; XM_002718526.1); human H2B (6), BamHI and NotI (histones, cDNA source: George Patterson, NIH; NM_021058.3); mouse mannosidase 2 (10), NheI and BamHI (cDNA source: Jennifer LippincottSchwartz, NIH; NM_008549.2); human pyruvate dehydrogenase (10), AgeI and NotI (PDHA1, cDNA source: Origene; NM_000284); yeast Lifeact (7), BamHI and NotI (cDNA source: IDT, Coralville, IA); human vimentin (7), BamHI and NotI (cDNA source: Robert Goldman, Northwestern University Medical School, Chicago, IL; NM_003380.3); human vascular epithelial cadherin (10), AgeI and NotI (cDNA source: Origene; NM_001795.3). Following digestion with the appropriate restriction enzymes and gel purification, the plasmids were ligated together with the similarly digested and gel-purified mEos4a or mEos4b cloning vector and amplified using a Plasmid Maxiprep kit (Qiagen, Valencia, CA). Optical Properties Spectroscopic characterization Absorption spectra of purified proteins in 50 mM tricine buffer (pH 8.2) were measured with an absorbance spectrometer (Lambda 35, Perkin-Elmer). mEos2 was additionally measured in 50 mM MOPS (pH 7.2) for comparison to literature values. All protein solutions were first centrifuged (10,000 RPM for 5 minutes) to remove scattering particles, with the supernatant used for measurements. Protein concentration was determined by UV absorbance at 280 nm, using a predicted extinction coefficient (in M-1cm-1) 280 = NTrp*5500 + NTyr*1490 + NCys*125 1,2. The extinction coefficient of the green form 505 was determined by the measured absorbance at 505 nm divided by the protein concentration. We also determined chromophore concentration for the green form using the alkali denaturation method 3-6, assuming a GFP-like extinction coefficient of 44,000 M-1cm-1 at 447 nm at pH 13.0. This method gave values for 505 that were consistently 27% higher than those found by protein concentration based on UV absorbance. Fluorescence correlation spectroscopy was additionally performed on several variants to determine chromophore concentration, using eGFP at pH 9.5 as a concentration reference, by the methods and instruments described in 7. The extinction coefficient of the red form 570 was found by measuring the initial change in optical density (OD) of the red and green forms of protein following irradiation with 385 nm light, assuming in an incremental time period the loss in concentration of green-form proteins by photoconversion equals the gain in concentration of red-form proteins. We accounted for the fact that the decrease in OD at 505 nm rides on top of the tail of the red-form absorbance peaked near 570 nm. Fluorescence excitation and emission spectra were recorded on either a microplate reader (Safire2, Tecan) or a luminescence spectrometer (LS55, Perkin-Elmer). Photoconversion on purified protein solutions was performed in cuvettes using a LED flood array (Loctite) operating at 385 nm (14 nm full-width at half-maximum) that delivered an intensity of 0.42 W/cm2 at the sample face. Quantum yields were measured in an integratingsphere spectrometer (Quantaurus, Hamamatsu) for both green-form and photoconverted red-form proteins (each solution 3 ml of 0.1 OD at peak absorbance). Photoswitching/Bleaching Time-course measurements were carried out on Tricine-buffered aqueous droplets of protein isolated by oil 8 on a widefield epi-illumination microscope (AxioImager.Z2, Zeiss) with a 20x 0.8 NA objective. Collinear laser excitation was provided to the microscope at 405 nm (Sharp laser diode, Thorlabs), 488 nm (Sapphire 488, Coherent) and 561 nm (Sapphire 561, Coherent) using beamcombining dichroics. In order to measure photoswitching, photoconversion, and bleaching, sequences of light pulses were delivered to the sample, cycling between 405/488 for the green form or between 405/561 for the red form, generated by computer-controlled shutters. Laser power was measured at the output of the objective, and the laser beam area and uniformity at the sample was imaged and measured using a CCD camera. We calculated the laser intensity from the measured laser power and beam area. Fluorescence collected by the objective was passed through appropriate dichroic/bandpass filter cubes (dichroic FF495-Di03 and bandpass 525/50 for green form, dichroic Di01-R405/488/561/635 and bandpass 625/90 for red form; all Semrock), and detected by a fiber-coupled avalanche photodiode APD (SPQM-AQRH14, Pacer), which sampled a sub-region of the illuminated protein droplet. The APD signal was summed every 40-msec time interval and stored on computer with its corresponding time point. Two-photon action spectra Two-photon spectra were obtained for 1 M protein solutions (as determined by alkali denaturation) in 50 mM tricine buffer, pH 8.2 using a Ti:sapphire laser (Chameleon Ultra II, Coherent) as excitation source on an inverted microscope (IX81, Olympus) with a 60x 1.2 NA water-immersion objective. The system, described in detail in 7, was set up to deliver a constant power (1 mW) at the sample across the spectrum of the laser (710 – 1080 nm). Fluorescence was collected by the objective, reflected off a dichroic, and passed through a bandpass filter (550/88 for green form, 625/90 for red form, Semrock) and detected by a fiber-coupled APD (SPQM-AQ14, Pacer). Reference spectra were obtained for fluorescein in 1 mM NaOH, pH 11.0 for the green form and tdTomato in 30 mM MOPS, 100 mM KCl, pH 7.2 for the red form. Reference values of action spectra or two-photon cross sections were taken from 9 for fluorescein and from 10 for tdTomato. PALM imaging of mEos4 variants fused to a variety of cellular proteins Sample Preparation for PALM imaging For PALM imaging, cells were seeded directly onto 35 mm, #0 thickness coverglass MatTek dishes (MatTek Corporation; Ashland, MA) approximately 24 hours prior to transfection. The culture medium was a 1:1 mixture of DMEM and Ham’s F12 supplemented with 12.5% cosmic calf serum, as described above. Transfection was performed using the Effectene transfection reagent (Qiagen) following the manufacturer’s instructions. Transfected cells were allowed to sit for approximately 24-48 hours prior to fixation, to ensure proper localization of the fusion construct. For keratin fusions, cells were fixed using -20oC methanol for 10 minutes and rehydrated in PBS. For α-tubulin fusions, cells were fixed in a 3% (w/v) PFA, 0.05% (v/v) glut, 0.3% (v/v) Triton X-100 solution in HEPES buffer, washed for approximately 5 minutes in a 0.1% solution of NaBH4 in PBS on ice, and rinsed several times in PBS. The remaining fusions were all fixed in 2% (w/v) PFA in PBS and washed twice with PBS containing 50 mM glycine. PALM Imaging The potential of mEos4a, mEos4b and mEos2 for PALM was compared by imaging fusions of each FP to a variety of cellular components (Additional Figs. 15). All PALM imaging parameters for fusion proteins were identical to those given for alpha-tubulin in the online Methods. 1 2 3 4 5 6 7 8 9 10 Gill, S. C. & von Hippel, P. H. Calculation of protein extinction coefficients from amino acid sequence data. Analytical biochemistry 182, 319-326 (1989). Pace, C. N., Vajdos, F., Fee, L., Grimsley, G. & Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein science : a publication of the Protein Society 4, 2411-2423, doi:10.1002/pro.5560041120 (1995). Adam, V. et al. Structural characterization of IrisFP, an optical highlighter undergoing multiple photo-induced transformations. Proceedings of the National Academy of Sciences of the United States of America 105, 1834318348, doi:Doi 10.1073/Pnas.0805949105 (2008). Gross, L. A., Baird, G. S., Hoffman, R. C., Baldridge, K. K. & Tsien, R. Y. The structure of the chromophore within DsRed, a red fluorescent protein from coral. Proceedings of the National Academy of Sciences of the United States of America 97, 11990-11995, doi:10.1073/pnas.97.22.11990 (2000). Ward, W. W. Biochemical and Physical Properties of Green Fluorescent Protein. Method Biochem Anal 47, 39-65 (2006). Ward, W. W., Cody, C. W., Hart, R. C. & Cormier, M. J. Spectrophotometric Identity of the Energy-Transfer Chromophores in Renilla and Aequorea Green-Fluorescent Proteins. Photochemistry and photobiology 31, 611-615, doi:Doi 10.1111/J.1751-1097.1980.Tb03755.X (1980). Mütze, J. et al. Excitation spectra and brightness optimization of two-photon excited probes. Biophysical journal 102, 934-944, doi:10.1016/j.bpj.2011.12.056 (2012). Kremers, G. J. & Piston, D. Photoconversion of purified fluorescent proteins and dual-probe optical highlighting in live cells. Journal of visualized experiments : JoVE, doi:10.3791/1995 (2010). Xu, C. & Webb, W. W. Measurement of two-photon excitation cross sections of molecular fluorophores with data from 690 to 1050 nm. J Opt Soc Am B 13, 481-491, doi:Doi 10.1364/Josab.13.000481 (1996). Drobizhev, M., Makarov, N. S., Tillo, S. E., Hughes, T. E. & Rebane, A. Twophoton absorption properties of fluorescent proteins. Nature Methods 8, 393399, doi:Doi 10.1038/Nmeth.1596 (2011). 11 12 13 14 15 16 17 18 19 Beckstead, J. H. A simple technique for preservation of fixation-sensitive antigens in paraffin-embedded tissues. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society 42, 1127-1134 (1994). Lomant, A. J. & Fairbanks, G. Chemical probes of extended biological structures: synthesis and properties of the cleavable protein cross-linking reagent [35S]dithiobis(succinimidyl propionate). Journal of molecular biology 104, 243-261 (1976). Mattson, G. et al. A practical approach to crosslinking. Molecular biology reports 17, 167-183 (1993). Tokumasu, F. & Dvorak, J. Development and application of quantum dots for immunocytochemistry of human erythrocytes. Journal of microscopy 211, 256-261 (2003). Hand, A. R. & Hassell, J. R. Tissue fixation with diimidoesters as an alternative to aldehydes. II. Cytochemical and biochemical studies of rat liver fixed with dimethylsuberimidate. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society 24, 1000-1011 (1976). Lakowicz, J. R. Principles of fluorescence spectroscopy. 3rd edn, (Springer, 2006). Beisker, W., Dolbeare, F. & Gray, J. W. An improved immunocytochemical procedure for high-sensitivity detection of incorporated bromodeoxyuridine. Cytometry 8, 235-239, doi:10.1002/cyto.990080218 (1987). Clancy, B. & Cauller, L. J. Reduction of background autofluorescence in brain sections following immersion in sodium borohydride. Journal of neuroscience methods 83, 97-102 (1998). Phillips, S. R., Wilson, L. J. & Borkman, R. F. Acrylamide and iodide fluorescence quenching as a structural probe of tryptophan microenvironment in bovine lens crystallins. Current eye research 5, 611-619 (1986).