View/Open - Lirias
advertisement

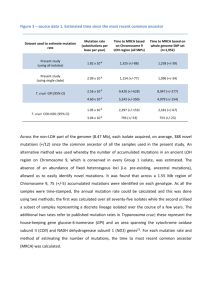
Ribosomopathies and the paradox of cellular hypo- to hyperproliferation Kim De Keersmaecker 1,2, Sergey O. Sulima 1,2 and Jonathan D. Dinman 3 1 Department of Oncology, KU Leuven, Leuven, Belgium 2 Center for the Biology of Disease, VIB, Leuven, Belgium 3 Department of Cell Biology and Molecular Genetics, University of Maryland, College Park MD, USA CORRESPONDENCE Kim De Keersmaecker, Campus Gasthuisberg O&N4, box 602, Herestraat 49, 3000 Leuven. Kim.DeKeersmaecker@cme.vib-kuleuven.be Jonathan D. Dinman, Microbiology Building #231, Department of Cell Biology and Molecular Genetics, University of Maryland, College Park MD 20742 USA. dinman@umd.edu WORD COUNTS: Abstract (max 200): 137 Main text (excl abstract, Figure legends and refs; max 4000): 3920 # references (max 100): 54 1 ABSTRACT Ribosomopathies are largely congenital diseases linked to defects in ribosomal proteins or biogenesis factors. Some of these disorders are characterized by hypoproliferative phenotypes such as bone marrow failure and anemia early in life, followed by elevated cancer risks later in life. This transition from hypo- to hyperproliferation presents an intriguing paradox in the field of hematology known as ‘Dameshek’s riddle.’ Recent cancer sequencing studies also revealed somatically acquired mutations and deletions in ribosomal proteins in T-cell acute lymphoblastic leukemia (T-ALL) and solid tumors, further extending the list of ribosomopathies and strengthening the association between ribosomal defects and oncogenesis. In this perspective, we summarize and comment on recent findings in the field of ribosomopathies. We explain how ribosomopathies may provide clues to help explain Dameshek’s paradox and highlight some of the open questions and challenges in the field. 2 INTRODUCTION Ribosomopathies are a collection of disorders of ribosome dysfunction characterized by a defect in ribosome biogenesis, typically due to a mutation in a ribosomal protein or biogenesis factor. They are rare diseases, with the most frequent having incidences in the range of 1 in 50 000 to 1 in 200 000 live births. These include Diamond-Blackfan Anemia (DBA) and 5qsyndrome that are associated with mutations in ribosomal proteins, as well as diseases linked to defects in ribosome biogenesis factors such as Schwachman-Diamond syndrome (SDS) and Treacher Collins syndrome (TCS). An example of a rarer ribosomopathy is X-linked dyskeratosis congenita. All of these diseases are characterized by distinct clinical phenotypes and patients present with one or several of a wide variety of developmental abnormalities including hematopoietic defects (e.g. anemia and other cytopenias), craniofacial malformations, short stature, mental and motor retardation. Although diverse, these phenotypes can all be categorized as cellular hypoproliferative defects. Intriguingly, some ribosomopathy patients are also at increased risk of developing leukemias and solid tumors, i.e. hyperproliferative cellular defects.1,2,3 For example: DBA is characterized by hypoproliferation phenotypes including macrocytic anemia, short stature, craniofacial defects and thumb abnormalities beginning early in life. In addition, these patients have been reported to have a 5-fold higher incidence of cancers, with the highest predisposition to colon cancer (36-fold higher incidence than in the general population), osteosarcoma (33-fold higher) and acute myeloid leukemia (28-fold higher).1 Although these associations with elevated cancer risks have been described in DBA, it is important to remark that this is a rare disease and that the number of cancer cases studied remains limited. Further studies demonstrating a causal link between ribosomal defects and elevated cancer risks in DBA are needed. Additionally, it is important to note that elevated cancer risks have not been demonstrated in all ribosomopathies, e.g. TCS has not been linked to any higher cancer predisposition. It is not our aim here to give an extensive overview of ribosomopathies or an in depth description of the exact clinical manifestations of the different diseases. Therefore, we refer to excellent reviews written by others.4,5,6 It is important to point out that ribosomopathies represent only a subset of congenital bone marrow failure syndromes: marrow failure and myelodysplasia with associated risk for transformation can also be caused by other classes of congenital and non-congenital defects such as mutations in genes involved in DNA repair (mutations in Fanconi anemia pathway),7 telomere regulation,8 epigenetics and splicing9. These are however not the subjects of this perspective. In this perspective, we summarize and comment on recent findings in the field of ribosomopathies. To begin, we highlight the rapid expansion of the ribosomopathy field due to the discovery of somatic mutations in ribosomal proteins in T-cell acute lymphoblastic leukemia (T-ALL) and other cancer samples. Next, we summarize the state-of-the-art of the molecular biology underlying the hypo- and hyperproliferative phenotypes in ribosomopathies and provide 3 a model that may partially explain the transition from one to the other (Dameshek’s riddle). Finally, we discuss some of the open questions and future perspectives in the field. 'TO BE' OR 'NOT TO BE' A RIBOSOMOPATHY: TOWARDS A DEFINITION The birth of the field of ribosomopathies dates back to 1999, when Draptchinskaia and colleagues described mutations in the ribosomal protein coding gene RPS19/eS19 (please note that the second nomenclature refers to the universal ribosomal protein nomenclature which was recently introduced to conform to atomic resolution ribosome structures from all three domains of life)10 in DBA.11 Since then, the list of ribosomopathies has continued to grow. One example is the recent discovery of congenital mutations in ribosomal protein SA (RPSA/uS2) in patients with isolated congenital asplenia.12 Another important expansion of the list of ribosomopathies comes from the recent application of unbiased next generation sequencing based mutation screening to cancer samples. Whereas previously all ribosomopathies (with the exception of the 5q- syndrome, linked to somatic loss of the RPS14/uS14 gene) have been associated with congenital genetic defects, genome wide sequencing efforts of cancer samples revealed that some of the genes associated with such congenital defects in ribosomopathies can also be targets of somatic mutation in cancers. For example, we recently identified inactivating mutations in the gene encoding ribosomal protein L5 (RPL5/uL18) in patients with T-ALL, some of which are identical to mutations described in DBA patients.13 Rare somatic mutations in T-ALL patients were also identified in RPL11/uL5, another gene that is affected in DBA.14 Interestingly, somatic defects in ribosomal proteins that have not been implicated in congenital ribosomopathies have now also been identified in T-ALL, gastric and ovarian cancer. These include somatic mutations in the RPL10/uL16 and RPL22/eL22 genes.13,15,16,17 Consistent with the observation that patients with congenital ribosomopathies are predisposed to develop various tumor types, the presence of somatic ribosome defects is emerging as a common occurrence in cancers.18 In the near future, completion of ongoing cancer sequencing projects will deepen our understanding of the spectrum of ribosomal mutations in cancer and may also identify ribosome biogenesis factors implicated in carcinogenesis. The discovery of somatic ribosome defects in cancer samples calls into question the current definition of ‘ribosomopathy’. Should the definition be limited to diseases and syndromes involving congenital mutations in ribosomal protein or biogenesis factor genes only, or should it be broadened to include somatically acquired mutations as well? Precedent for the latter definition may be found in 5q- syndrome. The observations that not all diseases typically accepted as ribosomopathies are associated with elevated cancer risks and that clinical hypoproliferative phenotypes have not been noted in cancers with somatic ribosome mutations suggests that the ‘ribosomopathies’ should be broadly defined. However, to err on the side of caution, we do think that a causal link between the ribosome defect and associated disease 4 should have been demonstrated before calling a disease a ribosomopathy. Therefore, we propose a conservative definition of ribosomopathies as ‘any disease associated with a mutation in a ribosomal protein or biogenesis factor impairing ribosome biogenesis in which a defect in ribosome biogenesis or function can be clearly linked to disease causality’. It is not clear at this point if a disease like cartilage hair hypoplasia falls under this definition. So far there has been no clear demonstration of a role for RNase MRP, the gene affected in cartilage hair hypoplasia, in ribosome biogenesis in humans. Moreover, it is currently unclear whether ribosome dysfunction is the cellular basis for the disorder. This definition also leaves ribosome mutant cancers in a gray zone: although loss of an RPL22/eL22 allele is able to accelerate tumor formation in a mouse model of T-cell leukemia,15 the causal role of other somatic ribosome mutations described in cancer remains to be shown. It is worth noting that somatic mutations in ribosomal proteins and biogenesis factors have only been described in a limited set of cancer samples to date. However, several cancer types are characterized by recurrent large deletions in which no tumor suppressor gene has yet been identified and which contain a ribosomal protein gene that is considered intrinsically ‘uninteresting.’ Our recent findings suggest that defects in ribosomal proteins and other synthesis factors should not be overlooked in cancer genotypes. Additionally, it has been clearly established by many research groups that protein synthesis appears deregulated in the majority, if not all, cancer samples. This is not surprising: this process plays such a central role in the cell that the signaling cascades and transcriptional regulators that are prominently altered in cancer also (de)regulate translation. Indeed, aberrations affecting the PI3K/AKT/mTOR pathway, the tumor suppressor protein p53 (TP53) pathway and the MYC transcription factor in cancer also impact cellular protein translation by deregulating expression of components of the translation machinery and by influencing the set of mRNAs which are most efficiently translated in the cell.19,20 However, whether altered translation in these instances is the cause of or consequence of cellular transformation is not clear. Therefore, we think it is not appropriate at this point to call such tumors ribosomopathies. HOW RIBOSOMOPATHIES CAN OFFER NEW INSIGHTS INTO DAMESHEK’S RIDDLE In a 1967 editorial, the founding editor-in-chief of Blood, William Dameshek, posed the following riddle: 'What do aplastic anemia, paroxysmal nocturnal hemoglobinuria (PNH) and 'hypoplastic' leukemia have in common?'.21 Here, Dameshek described the intriguing paradox in which patients who initially develop a hypoproliferative disease such as aplastic anemia tend to be at higher risk of hyperproliferative diseases such as PNH (clonal expansion of red cells) or acute leukemia (clonal expansion of white cells) later in life. Although not explicitly described by Dameshek, this observation of a class of diseases characterized in the early stages by a hypoproliferative disorder followed by the propensity to develop into a hyperproliferative disease fits the clinical presentation of certain ribosomopathies. As such, Dameshek’s riddle 5 may be more broadly applicable than initially postulated. The challenge is to explain this paradox. Dameshek proposed that a sufficiently severe insult to the marrow - whether chemical, ionizing radiation, viral or by a congenital defect - may result in a variable degree of injury with a variable degree of hypoplasia. In some cases, abnormal clones of abnormal leukocytes (hypoplastic leukemia) or red cells (PNH) could conceivably arise during the process of repair.21 In other words, Dameshek proposed that an initial insult first results in hypoproliferative disease, and that something involved in the process of its repair provides a subset of cells with the capacity to proliferate excessively. This idea was conceived in 1967, before the molecular genetics era. Remarkably, his model remains consistent with the models that are currently being proposed to explain Dameshek’s riddle. The differences lie in the depth of our understanding of the molecular nature of the 'initial insult', the identities of the signaling cascades that lead to impaired proliferation, and the nature of the molecular 'repair processes' that may underlie the transition to hyperproliferation. As discussed above, the initial insult occurring in ribosomopathies are defects in ribosomal protein genes and biogenesis factors. Ribosome biogenesis is a complex multistage process.22 Given the central role of the ribosome in the genetic program, there is strong pressure in cells to identify and remove malfunctioning ribosomes before they are released into the cytoplasmic pool of actively translating ribosomes. It is becoming clear that this process involves quality control mechanisms that operate at each step of the process.23,24 This has two consequences. The first is that cells may not produce enough functional ribosomes to support their metabolic demands. This is particularly germane to hematopoietic cells, whose normally high proliferative rates require large amounts of ribosomes. This alone may account for cellular hypoproliferation. In addition, too few functional ribosomes due to insufficient quantities of one or more ribosomal proteins may also result in the release of excess free ribosomal proteins into the cytoplasm. This is a molecular stress signal. The most well-documented research in this area focuses on cytoplasmic release of RPL5/uL18 and RPL11/uL5. These have been shown to bind to and inhibit the murine double minute 2 protein (MDM2, or HDM2 in humans), a ubiquitin ligase that directs degradation of the TP53 protein. HDM2 inhibition results in TP53 stabilization, initiating a series of cellular signaling cascades leading to cell cycle arrest and apoptosis, adding to cellular hypoproliferation.25,26,27,28,29,30 This research only represents the tip of the iceberg: TP53 independent cell cycle arrest and apoptosis mechanisms have been described that may also contribute to the ribosomopathic-stress induced hypoproliferation.31,32,33,34 Although the human ribosome consists of 81 ribosomal proteins, only a fraction of these have been described as mutated or deleted in ribosomopathies. Similarly, only a restricted set of biogenesis factors have been implicated in ribosomopathies to date.4,5 This is most likely explained by the fact that mutation or deletion of many ribosomal proteins and biogenesis factors would be incompatible with cell survival and affected cells would thus not live long 6 enough to produce the ribosomopathic phenotype. Indeed, the ribosomal proteins currently implicated in ribosomopathies encode proteins located on the cytoplasmic/solvent-accessible surfaces of the ribosome.35 None of these proteins stabilize interdomain rRNA interactions, nor are they located in regions of the ribosome that directly mediate mRNA decoding or peptidyl transfer, leaving ribosomopathic ribosomes functional enough to support viability. Two typical trans-acting ribosomopathogenic factors, Shwachman–Bodian–Diamond syndrome protein (SBDS) and dyskerin (DKC1), are also consistent with this idea: SBDS is involved in the late, cytoplasmic quality control of pre-60S subunits,24 and DKC1 catalyzes pseudouridylation of rRNA. Although rRNA modifications occur early during rRNA transcription, these are thought to merely “fine-tune” ribosome structure rather than playing central roles in domain folding.36,37,38 The next issue concerns the nature of the 'repair process' in Dameshek’s model. Not surprisingly, this is less well understood as it is more complex. To approach this, we have turned to molecular evolutionary theory. As described above, the original mutation results in too few cells available to perform their physiological function, e.g. production of red blood cells. From an evolutionary standpoint, this places pressure on the dwindling population of hematopoietic stem cells, selecting for mutations that enable cells to produce enough ribosomes to meet their protein synthetic requirements. One such class of mutants are those that repair the original defect (revertants). A second class, referred to as second site suppressors, are mutations that either inactivate or bypass the ribosome biogenesis quality control apparatus. Cells harboring this class of mutations would produce enough ribosomes to satisfy their translational requirements, enabling them to out-compete those having only the original mutation. While this solves the hypoproliferation problem, the tradeoff is that these cells must utilize defective ribosomes. Emerging research indicates that these ribosomes have specific defects in translational fidelity, i.e. their ability to faithfully translate the genetic code. Indeed, our laboratories have begun to identify a causal chain of events detailing how ribosomopathy associated defects such as RPL10/uL16 R98S or Cbf5p-D95A (a catalytically impaired mutant of Cbf5p, the yeast homologue of DKC1) cause subtle changes in ribosome structure that alter the basic biochemistry of mutant ribosomes, and how this in turn affects specific aspects of translational fidelity, altering the expression of specific sets of genes. 35,38 For RPL10/uL16 R98S, we also showed that these changes in gene expression confer new stresses on this population of cells, hastening the selection for additional compensatory mutations that eventually result in cellular immortalization, i.e. hyperproliferation. 35 This is a classic ‘deal with the devil’: the process that initially enables cells to fulfill their physiological function eventually leads to their uncontrolled growth and cancer. The current challenge is to identify the critical second site suppressors. Once identified, rational strategies can be designed to reverse their effects. The ideal samples would be tumors collected from patients that developed a ribosome defect associated malignancy. Such patient 7 samples are however scarce because, fortunately, not all patients develop malignancies. Moreover, cancer samples are often characterized by genetic instability and accumulation of defects, making it difficult to pinpoint the exact nature of the original hypoproliferation suppressor mutation. This is where the molecular genetics of model organisms becomes a powerful tool. Recent work from our laboratories provides insight into the molecular nature of the 'repair process'. This began with identification of a recurrent arginine to serine mutation (R98S) in ribosomal protein RPL10/uL16 in T-ALL patient cells. Intriguingly, while T-ALL represents a hyperproliferative disease, introduction of this mutation into yeast and mammalian cells impairs ribosome biogenesis and cellular proliferation causing the hypoproliferative phenotype.13 In depth analysis of the RPL10/uL16 R98S mutant using the yeast genetic model revealed that this mutation does not completely inhibit ribosome biogenesis: this is why they are alive, albeit barely. Importantly, allowing cells to grow over many generations selected for a population of cells with normal growth rates. Genetic analysis revealed that this was due to acquisition of a compensatory mutation in a ribosome biogenesis factor that bypassed a critical quality control checkpoint. Importantly, while this second site mutation rescued the hypoproliferative defect, it did not correct the underlying structural, biochemical and translational fidelity defects and altered gene expression profiles conferred by the original RPL10/uL16 R98S mutation (Figure 1A, B).35 While we have not been able to identify analogous mutations in biogenesis factors in RPL10/uL16 R98S positive T-ALL samples, this may be due to the paucity of knowledge pertaining to human biogenesis factors, i.e. mammalian ribosome biogenesis factors are still poorly defined. Additionally, yeast and human cells are sufficiently different that mutations in alternative classes of genes may be required to rescue human cell proliferation defects. For example, human cells may mutate components of the TP53 pathway or of the other pathways that initiate hypoproliferation upon ribosome biogenesis-induced stress (Figure 1C). It would be highly interesting to perform polysome profiling on non-cancerous versus cancerous tissue from patients with a congenital ribosome defect to determine whether a compensatory mutation was acquired that corrects the biogenesis defect (Figure 1B) or whether the biogenesis defect is still present, as predicted by the model shown in Figure 1C. Zebrafish are also becoming an attractive model to study ribosome mutation associated cancers. Seventeen zebrafish lines have been described harboring heterozygous inactivation of different ribosomal protein genes that are predisposed to develop malignancies. These tumors have much in common with those observed in Tp53 null zebrafish, and tumors induced by ribosomal protein haploinsufficiency showed a specific translation defect impairing Tp53 protein production.39 Unfortunately, a hypoproliferative phenotype has not been described in these fish, and thus it remains unclear whether or not defective Tp53 translation can actually cause a switch from a hypo- to a hyperproliferative phenotype in this model system. A mouse model on RPL22/eL22 haploinsufficiency supports the idea that a ribosomal protein defect may contribute to a hyperproliferative phenotype provided that additional mutations are present. 8 RPL22/eL22 has been described as a target for heterozygous somatic mutations and deletions in T-ALL and in microsatellite instability-positive gastric and endometrial cancer.15,40,17 Whereas Rpl22/eL22 +/- mice do not develop any malignancies within 25 weeks, crossing of these mice to a strain with T-cell specific expression of myristoylated Akt2 (MyrAkt2) accelerates the latency of T-cell lymphoma development from 19 weeks (MyrAkt2- Rpl22/eL22+/+) to 11 weeks (MyrAkt2- Rpl22/eL22+/-).15 Although very interesting, these data still do not identify the nature of the extra mutations that human cancer cells acquire to be transformed in the context of a ribosomal defect. It is also worth remarking that although loss of 1 copy of Rpl22/eL22 is sufficient to accelerate T-cell lymphoma development in a MyrAkt2 background, loss of both Rpl22/eL22 copies is needed to observe a hyperproliferative phenotype of arrested development of alphabeta-lineage T cells at the beta-selection checkpoint.15,41 FUTURE PERSPECTIVES AND CONCLUSIONS A long list of genes associated with ribosomopathies has been generated over the past three decades. However, it is clear that we are still far from the goal of a complete understanding of the pathogenesis of ribosomopathies. Essential information regarding the cellular changes associated with ribosomal mutations remains to be discovered. While mRNA expression studies may provide certain clues,42,43,44,45 given that protein translation is the central function of ribosomes, it may be essential to complement these studies by a detailed analysis of the effects on protein production. This necessity is illustrated by the recent finding that in a substantial fraction of DBA cases, one consequence of the ribosomal protein mutations is failure of translation of the transcription factor GATA-1, an essential factor for development of erythropoiesis beyond the CFU-E stage. This may be because the long form of GATA-1 is difficult to translate and is impaired in DBA cells.46 These translational defects of GATA-1 were found because of the rarity of GATA-1 mutations in DBA. However, additional translation defects in DBA and other ribosomopathies have probably not been discovered because of a lack of thorough comparative proteomic studies of normal versus ribosomopathic cells. Unfortunately, this is a classic example of a problem waiting for a technological advance; proteomics technologies are still evolving and analyzing an entire proteome of human cells is not yet within reach of research labs. At this juncture, sequencing of polysome associated RNA undergoing translation is an option.47,48,49 The best approximation at this point is ribosome profiling technology, which allows not only identification of ribosome protected (= translated) mRNA fragments but also analysis of alternative start codon usage, read-through beyond stop codons and of translational kinetics.50,51 Additionally, although the need for cooperating mutations appears to be clear, questions remain regarding the nature of these mutations, the moment when they are acquired and the amount required to stimulate hyperproliferation. One attractive possibility accounting for the varying penetrance of cancers in different ribosomopathy patients may lie in their genetic backgrounds, i.e. cooperating mutations may be inherited as allelic variants that are normally maintained in the population. 9 Yet another intriguing paradox regarding ribosomopathies is the issue of phenotypic tissue specificity: why are such a wide spectrum of clinical presentations with typical tissue-specific pathologies observed for each disease, despite the fact that they all share a common defect in the same biochemical process? While patients with congenital ribosomopathies are born with the same ribosome defect in each cell of their body, not all tissues are affected and only particular disease symptoms are developed.5 Interestingly, although mutations in several ribosomal proteins and biogenesis factors give rise to anemia in different ribosomopathies, each disease also exhibits additional unique features. Even within the same ribosomopathy, mutations in different ribosomal protein genes promote different types of birth defects. For instance, most patients with RPL5/uL18 mutations have a cleft palate, whereas most individuals with RPL11/uL5 mutations do not have any craniofacial defects.52 As such, patients with genetic disorders that are linked to mutations in ribosomal proteins show remarkably specific phenotypes, suggesting that ribosomal proteins have unique functions in different tissues. Dissecting the molecular and biochemical defects of different types of mutant ribosomes can contribute to the exciting emerging field of ‘specialized ribosomes’ – ribosomes whose specificity may be controlled and fine-tuned by tissue-specific and developmentally regulated signals. Alternatively, essential tissue-specific roles of ribosomal proteins outside of the ribosome (extra-ribosomal functions) may explain the tissue-specific phenotypes. Indeed, there is a growing body of evidence that ribosomal proteins do not only function in translation and that they can also regulate transcription and enzyme activity in the cell.53 In conclusion, many potential explanations for cellular transformation exist and are important subjects for investigation. We speculate that the ribosomal protein and biogenesis mutants in ribosomopathies allow the ribosome to become mature enough to support some degree of translation, yet still cause sufficient perturbations of ribosomal function to effect the changes in gene expression associated with ribosomopathic phenotypes. Indeed, we showed that yeast cells expressing a T-ALL associated RPL10/uL16 R98S mutant or a Cbf5p-D95A mutant promote specific defects in translation fidelity.35,38 Based on our work on RPL10/uL16 R98S, 35 we also hypothesize that compensatory mutations might be likely drivers of transformation, and are hence particularly important because they allow bypass of the cellular proliferation defects due to defective ribosome biogenesis. One class of compensatory mutations comprises mutant biogenesis factors which suppress the assembly but not the functional defects of mutant ribosomes. This leads to increased cellular utilization of defective ribosomes with altered fidelity. Additional experimental work is needed to validate this model in other systems and for other ribosomal defects. Moreover, while bypassing the ribosomal assembly quality control through mutated biogenesis factors may provide one window to oncogenesis, it is likely that other classes of oncogenic secondary mutations exist. Indeed it is becoming clear that cooperating secondary development.54 mutations/suppressors are frequently required for disease Transformation merely represents an endpoint that is likely made more 10 accessible by evolutionary selection for secondary mutations in a variety of cellular pathways. Determining the nature of the pathways and the mutations is an exciting and promising field for investigation. 11 ACKOWLEDGEMENTS KDK receives a postdoctoral fellowship of FWO Vlaanderen. SOS is recipient of an EMBO longterm postdoctoral fellowship. This work was partially supported by a grant from the National Institutes of Health to JDD (R01 HL119438) and by an FWO grant (G.0840.13), a Stichting Tegen Kanker (F25) and an ERC starting grant (GA 334946) to KDK. AUTHORSHIP CONTRIBUTIONS KDK, SOS and JDD wrote and edited the manuscript. DISCLOSURE OF CONFLICTS OF INTEREST The authors declare no competing financial interests REFERENCES 1.Vlachos A, Rosenberg PS, Atsidaftos E, Alter BP, Lipton JM. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood. 2012;119(16):3815-9. 2.Rosenberg PS, Alter BP, Bolyard AA, et al. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. 2006;107(12):4628-35. 3.Alter BP, Giri N, Savage SA, et al. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br. J. Haematol. 2010;150(2):179-88. 4.Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115(16):3196-205. 5.Armistead J, Triggs-Raine B. Diverse diseases from a ubiquitous process: the ribosomopathy paradox. FEBS letters. 2014;588(9):1491-500. 6.Ruggero D, Shimamura A. Marrow failure: a window into ribosome biology. Blood. 2014;124(18):2784-2792. 7.Longerich S, Li J, Xiong Y, Sung P, Kupfer GM. Stress and DNA repair biology of the Fanconi anemia pathway. Blood. 2014;124(18):2812-2819. 8.Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124(18):2775-2783. 9.Bejar R, Steensma DP. Recent developments in myelodysplastic syndromes. Blood. 2014;124(18):2793-2803. 10.Ban N, Beckmann R, Cate JHD, et al. A new system for naming ribosomal proteins. Curr. Opin. Struct. Biol. 2014;24:165-9. 11.Draptchinskaia N, Gustavsson P, Andersson B, et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 1999;21(2):169-75. 12.Bolze A, Mahlaoui N, Byun M, et al. Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science. 2013;340(6135):976-8. 12 13.De Keersmaecker K, Atak ZK, Li N, et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2013;45(2):186-190. 14.Tzoneva G, Perez-Garcia A, Carpenter Z, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nature medicine. 2013;19(3):368-71. 15.Rao S, Lee S, Gutierrez A, et al. Inactivation of ribosomal protein L22 promotes transformation by induction of the stemness factor, Lin28B. Blood. 2012;120(18):3764-73. 16.Wang K, Kan J, Yuen ST, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 2011;43(12):1219-23. 17.Novetsky AP, Zighelboim I, Thompson DM, et al. Frequent mutations in the RPL22 gene and its clinical and functional implications. Gynecol. Oncol. 2012;128(3):470-4. 18.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013;502(7471):333-9. 19.Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nature Reviews Cancer. 2003;3(3):179-192. 20.Stumpf CR, Ruggero D. The cancerous translation apparatus. Current opinion in genetics & development. 2011;21(4):474-83. 21.Dameshek W. Riddle: what do aplastic anemia, paroxysmal nocturnal hemoglobinuria (PNH) and “hypoplastic” leukemia have in common? Blood. 1967;30(2):251-254. 22.Strunk BS, Karbstein K. Powering through ribosome assembly. RNA. 2009;15(12):2083104. 23.Matsuo Y, Granneman S, Thoms M, et al. Coupled GTPase and remodelling ATPase activities form a checkpoint for ribosome export. Nature 2013;505(7481):112-6. 24.Lo K, Li Z, Bussiere C, et al. Defining the pathway of cytoplasmic maturation of the 60S ribosomal subunit. Molecular cell. 2010;39(2):196-208. 25.Dai M, Lu H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J. Biol. Chem. 2004;279(43):44475-82. 26.Marechal V, Elenbaas B, Piette J, Nicolas JC, Levine AJ. The ribosomal L5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol. Cell. Biol. 1994;14(11):7414-20. 27.Zhang Y, Wolf GW, Bhat K, et al. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell. Biol. 2003;23(23):8902-12. 28.Lohrum MAE, Ludwig RL, Kubbutat MHG, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3(6):577-87. 29.Bursać S, Brdovčak MC, Pfannkuchen M, et al. Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. Proc. Natl. Acad. Sci. U.S.A. 2012;109(50):20467-72. 30.Bursac S, Brdovcak MC, Donati G, Volarevic S. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim. Biophys. Acta. 2014;1842(6):817-830. 13 31.Donati G, Montanaro L, Derenzini M. Ribosome biogenesis and control of cell proliferation: p53 is not alone. Cancer research. 2012;72(7):1602-7. 32.Narla A, Payne EM, Abayasekara N, et al. L-Leucine improves the anaemia in models of Diamond Blackfan anaemia and the 5q- syndrome in a TP53-independent way. Br. J. Haematol. 2014;167(4):524-528. 33.Singh SA, Goldberg TA, Henson AL, et al. p53-Independent cell cycle and erythroid differentiation defects in murine embryonic stem cells haploinsufficient for Diamond Blackfan anemia-proteins: RPS19 versus RPL5. PloS one. 2014;9(2):e89098. 34.Heijnen HF, van Wijk R, Pereboom TC, et al. Ribosomal Protein Mutations Induce Autophagy through S6 Kinase Inhibition of the Insulin Pathway. PLoS genetics. 2014;10(5):e1004371. 35.Sulima SO, Patchett S, Advani VM, et al. Bypass of the pre-60S ribosomal quality control as a pathway to oncogenesis. Proc. Natl. Acad. Sci. U.S.A. 2014;111(15):5640-5645. 36.Decatur WA, Fournier MJ. rRNA modifications and ribosome function. Trends in biochemical sciences. 2002;27(7):344-51. 37.Baxter-Roshek JL, Petrov AN, Dinman JD. Optimization of ribosome structure and function by rRNA base modification. PloS one. 2007;2(1):e174. 38.Jack K, Bellodi C, Landry DM, et al. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Molecular cell. 2011;44(4):660-6. 39.MacInnes AW, Amsterdam A, Whittaker CA, Hopkins N, Lees JA. Loss of p53 synthesis in zebrafish tumors with ribosomal protein gene mutations. Proc. Natl. Acad. Sci. U.S.A. 2008;105(30):10408-13. 40.Nagarajan N, Bertrand D, Hillmer AM, et al. Whole-genome reconstruction and mutational signatures in gastric cancer. Genome Biol. 2012;13(12):R115. 41.Anderson SJ, Lauritsen JPH, Hartman MG, et al. Ablation of ribosomal protein L22 selectively impairs alphabeta T cell development by activation of a p53-dependent checkpoint. Immunity. 2007;26(6):759-72. 42.Ebert BL, Pretz J, Bosco J, et al. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature. 2008;451(7176):335-9. 43.Aspesi A, Pavesi E, Robotti E, et al. Dissecting the transcriptional phenotype of ribosomal protein deficiency: implications for Diamond-Blackfan Anemia. Gene. 2014;545(2):282-9. 44.Devlin EE, Dacosta L, Mohandas N, Elliott G, Bodine DM. A transgenic mouse model demonstrates a dominant negative effect of a point mutation in the RPS19 gene associated with Diamond-Blackfan anemia. Blood. 2010;116(15):2826-35. 45.Rujkijyanont P, Adams S, Beyene J, Dror Y. Bone marrow cells from patients with Shwachman-Diamond syndrome abnormally express genes involved in ribosome biogenesis and RNA processing. Br. J. Haematol. 2009;145(6):806-15. 46.Ludwig LS, Gazda HT, Eng JC, et al. Altered translation of GATA1 in Diamond-Blackfan anemia. Nature medicine. 2014;20(7):748-53. 14 47.Horos R, Ijspeert H, Pospisilova D, et al. Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood. 2011;119(1):262-72. 48.Pereboom TC, Bondt A, Pallaki P, et al. Translation of branched-chain aminotransferase-1 transcripts is impaired in cells haploinsufficient for ribosomal protein genes. Exp. Hematol. 2014;42(5):394-403.e4. 49.Hesling C, Oliveira CC, Castilho BA, Zanchin NIT. The Shwachman-Bodian-Diamond syndrome associated protein interacts with HsNip7 and its down-regulation affects gene expression at the transcriptional and translational levels. Experimental cell research. 2007;313(20):4180-95. 50.Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nature protocols. 2012;7(8):1534-50. 51.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147(4):789-802. 52.Gazda HT, Sheen MR, Vlachos A, et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am. J. Hum. Genet. 2008;83(6):769-80. 53.Lindström MS. Emerging functions of ribosomal proteins in gene-specific transcription and translation. Biochemical and biophysical research communications. 2008;379(2):167-70. 54.Ishimura R, Nagy G, Dotu I, et al. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014;345(6195):455-459. FIGURE LEGENDS Figure 1. Model explaining transition from hypo- to hyperproliferation phenotypes in ribosomopathies. (A) In the initial phase of the disease, a mutation in a ribosomal protein or ribosome biogenesis factor (symbolized by the red star on the 60S subunit) impairs proper ribosome biogenesis, resulting in lower concentrations of assembled ribosomes. This ribosome biogenesis defect impairs proper proliferation of the cells by activating the TP53 pathway and/or by TP53 independent mechanisms. At this stage, functional ribosomes are still assembled to a certain extent. These ribosomes are however intrinsically defective and may induce translational changes/shifts in the cells. (B + C) The impaired proliferation imposes strong pressure on cell populations, selecting for cells that acquire a compensatory mutation rescuing the impaired proliferation. The nature of this compensatory mutation is currently unclear. Cells may acquire a mutation in a biogenesis factor rescuing the biogenesis defect (B). Alternatively, the signaling pathways inducing the proliferation impairment upon a biogenesis defect (TP53 or other) may be crippled by a compensatory mutation (C). In both scenarios, after acquiring the compensatory mutation, defective ribosomes would still be formed that alter the translational capacity/fidelity leading to the cell obtaining a clonal advantage over other cells. 15 However, these ribosomes are intrinsically defective, leading to altered gene expression programs. More speculative parts in the figure are indicated by dashed arrows. 16 17