Acknowledgements
Dr. Aurora Burds Connor for providing mouse embryos, lab protocols and instruction
Scientific Director, Mouse ES Cell & Transgenic Facility
Koch Institute for Integrative Cancer Research at MIT
Room 76-195, phone x2-3658
http://ki.mit.edu/sbc/escell
Lindsay Julien for ordering and donating materials, and for protocols for TNF-alpha lab
Saeij Lab manager
Department of Biology, 68-264D
Massachusetts Institute of Technology
77 Massachusetts Avenue, Cambridge, Ma 02139
Yisi Lu and Dr. Maria Ines Baptisa (Tyle Jacks lab)
Koch Institute for Integrative cancer Research
for providing mouse cell culture, media and supplies.
Megan Krench
Troy Littleton’s Lab
Department of Brain and Cognitive Sciences
For providing insect cells and culture media
Danielle Feldman for providing IPS and differentiated neuronal cells
Mriganka Sur’s lab
Department of Brain and Cognitive Sciences
Dr. Joahnna Scheuermann for providing cultures of differentiated cardiac cell
Laurie Boyer’s lab
Department of Biology
Dr. Emily Rosowski for protocols for TNF-alpha experiments
Saeij Lab manager
Department of Biology
High school interns Anahita Shahrokh and Alev Cansever for helping set up the labs
TW 2013
2
Welcome to the 2013 annual MIT summer workshop
for high school biology teachers!
The workshop will consist of lectures and laboratory exercises focused on the use of cell and
tissue culture to study development, diseases and molecular pathways.
The lectures will be held daily from 10:45 to noon followed by a buffet lunch from noon
to 1 PM.
All lectures will be in room 68-180 in the biology building
The Labs will be held daily from 9:00 AM to 10:30 AM and from 1 PM to 4:30 PM.
All labs will be in room 68-077.
Please arrive in room 68-180 by 8:30 (as early as 8:00 AM). We will begin at 8:30 AM
with an introduction and discussion of the agenda for the day, and will move to room 68077 to start the experiments by 9:00 AM.
The lectures will cover examples of the use of tissue culture in stem cell research and
infectious diseases, the immune response, and topics that include current approaches to
cancer treatment and personalized medicine.
The labs will introduce participants to the basics technique of cell culture, including how
to start cell culture from mouse embryos, how to count cells, determine cell viability and
how to measure cell proliferation (division). You will be ale to test the effect of various
chemicals on cell growth and viability that is similar to how researchers and
pharmaceutical companies test for novel drugs , and observe the immune response to
disease-causing pathogens.
TW 2013
3
LABORATORY SAFETY RULES
NOTE:
Wear a lab coat at all time in the lab.
Wear gloves when handling chemicals
Dispose of all cell cultures and contaminated liquids in 10% bleach.
Dispose of all tips, and sharps in appropriate grey containers
Keep benches clean and free of personal belongings at all time
Wipe bench with 70% ethanol before and after each experiments.
Wash hands with soap or alcohol-based hand sanitizers
Dispose of all pipettes and reagents in appropriate containers.
Keep written records of any observation and any deviation from protocols.
Record type of equipment used (name, brand, model) and name of supplier.
ABSOLUTELY NO EATING OR DRINKING IN THE LAB!
LABORATORY SCHEDULE: ALWAYS IN ROOM 68-077 (basement)
Morning session:
Afternoon sessions
9:00 AM to 10:30 AM
1:00 PM to 4:30 PM
You will be working in 8 teams of 2.
The morning laboratory sessions will include:
Introduction to afternoon experiments
Setting up for the afternoon experiments
Reviewing the previous days experiments
Analyzing the data obtained from the previous days experiments
LABORATORY SCHEDULE
The week at a glance
TW 2013
4
Day 1: MONDAY JULY 15
MORNING
Overview of experiments, lab tour and safety guidelines/training
Team assignments
Learn how to work in tissue culture hood using sterile technique
Observe cells under the inverted microscope
AFTERNOON
Generate mouse embryo fibroblasts (MEFs) with Dr. Aurora Burds Connor
Learn how to count and split cells in preparation for Tuesday’s experiment
Day 2: TUESDAY JULY 16
MORNING:
Count and plate insect S2 cells for proliferation assay using the Trypan blue exclusion
assay
Observe the progression of the cultured MEFs started on day 1
AFTERNOON
Add TNF-alpha to cells, block overnight
Observe the progression of cultured MEFs
Day 3: WEDNESDAY JULY 17
MORNING
Observe the progression of cultured MEFs
Feed MEFs cultures
Treat insect S2 cells with cytotoxic doses of household chemicals
Start antibody staining procedure on TNF-alpha-treated cells: add primary Ab (anti-p65)
AFTERNOON
Complete antibody procedure on TNF-alpha-treated cells
Count insect S2 cells from Tuesday to determine rate of cell proliferation
Observe undifferentiated vs. induced neuronal cell cultures
Observe transfection of insect S2 cells with wild-type and mutant constructs
Day 4: THURSDAY JULY 18 MORNING
Split cultured MEFs
TW 2013
5
Observe the cellular localization of NF-kappaB with and without TNF-alpha treatment
AFTERNOON
Count insect S2 cells from Wednesday to determine rate of cell proliferation
Observe transfected insect S2 cells, and record any difference between the transfections
Observe stem cells: undifferentiated vs. differentiated cardiac cells
Day 5: FRIDAY JULY 19
MORNING:
Observe passaged MEFs
Observe transfected insect S2cells, record any difference between the transfections.
Clean up, discard properly all cell cultures
AFTERNOON:
Project presentations
Review and Discussion of experiments and results
Curriculum presentation
TW 2013
6
A quick overview of the experiments
Generating primary mouse embryo fibroblast (MEF) cultures
On the first day, each of you will generate your own primary culture of MEF cells or neurons
from a mouse embryo at embryonic day (E) 12.5 or E13.5; this means 12.5 or 13.5 days after
fertilization. Dead mouse embryos will be provided by Dr. Aurora Burds Connor the director
of the Mouse ES cell and transgenic facility in the Koch Institute. She will also demonstrate
how to process the embryos:
You will remove internal organs (anything red or pink), mince the remaining tissues with a
razor blade in media containing the trypsin enzyme, add growth media to the minced embryo
and plate the resulting liquid on tissue culture plates.
You will observe the progression of the cells over the next few days and record your
observations (cell morphology, cell density,..) When the cells have divided enough so that
there are little free spaces between them, they are considered “confluent” and are ready to be
split, which means you will detach the cells from the dish with trypsin and plate them onto new
tissue culture dishes; the cells will attach and begin dividing again.
Cell Proliferation assay using “S2 cells” derived from Drosophila melanogaster
Each team will receive a plate of confluent insect cells, count the number of live cells and then
split the cells. You will determine the rate of growth (proliferation) by counting the number of
live cells one and two days after splitting them.
To illustrate that cells can be very sensitive to environmental factors, you will treat some of the
cells with common chemicals such as salt, vinegar, and alcohol and you will determine the
effect these compounds have on cell proliferation and viability.
TNF-alpha treatment of cultured mouse cells, and determination of NF-kappaB protein
localization.
You will obtain 2-well glass chamber slides that contain mouse cells. You will treat one of the
wells with tumor necrosis factor-alpha (TNF-alpha), a cytokine that is secreted by macrophage
cells of the immune system, and leave the second well untreated for comparison (negative
control). The treatment mimics an immune response and will demonstrate how the presence of
TNF-alpha in the media affects cellular signaling. This effect on signaling can be visualized by
performing antibody staining on the cells to determine the location of the transcription factor
NF-kappaB, which is activated by TNF-alpha and regulates transcription of a set of specific
genes, including some that protect against induced cell death.
TW 2013
7
Protein expression in S2 cells
You will be introduced to the basic technique of introducing DNA into cultured cells
(transfection) for the purpose of protein expression.
To illustrate the utility of cell culture in understanding disease pathology, the normal (wildtype) and a mutant version of the huntingtin gene (htt) will be expressed in the S2 cells. The
htt gene encodes the huntingtin protein, In the neurological disorder Huntington’s Disease the
htt gene has an expanded repeat of CAG trinucleotides that lead to an expanded polyglutamine
repeat in the protein. This seems to lead to misfolding of the protein and clumping.
The normal function of the huntingtin protein is unclear but studies of samples from
Huntington’s Disease patients suggests that the mutant protein forms aggregates that disrupt
the normal functions of neurons and leads to degeneration of striatum neurons.
You will observe cells expressing the wild-type Huntingtin protein and compare it with cells
expressing a patient-associated mutation at 24 and 48 hours post-transfection and try to look for
a distinct phenotype associated with the disease.
For more information about the huntingtin protein see:
https://www.stanford.edu/group/hopes/cgi-bin/wordpress/2011/07/normal-huntingtin-andhuntingtons-disease/
TW 2013
8
INTRODUCTION
Mouse embryo fibroblast (MEF) culture
Only some type of cells can survive and multiply in vitro in dishes. Neurons and fibroblast
are two examples. Fibroblasts are found in connective tissue. They provide structural support
in the body and secrete proteins that form the extracellular matrix, which can be thought of as
a lattice upon which other cells can adhere. Fibroblasts can be cultured from mouse embryos
that are harvested 12.5 to 14.5 days after fertilization (embryonic day (E)12.5 and E14.5) (see
embryonic stages on the next page).
To prepare a culture of MEFs, mouse embryos are decapitated to remove the head (and the
neurons). Internal organs are also removed, and the remainder of the embryo is minced and
treated with the enzyme trypsin that cleaves proteins involved in cell-cell adhesion, to help
dissociate the cells from one another. The individual cells can attach on surfaces and grow if
provided with proper nutrient and conditions that can mimic the natural cellular environment
in the body.
MEFs isolated from embryos are extremely useful in the laboratory. They can be manipulated for
the study of protein expression and function in vitro. MEFs isolated from mouse mutant that carry a
genetic disease can provide insight into disease pathology when compared with wild-type MEFs in
cellular assays. In addition MEFs are quite useful in stem cell research. They secrete extracellular
factors and provide support and nutrients to the stem cells. Human embryonic stem cells (hESCs)
are generally maintained on a layer of inactive (irradiated) mouse embryonic fibroblast (MEF)
feeder cells to keep them healthy, happy and undifferentiated. The irradiation prevents the MEFs
from dividing and proliferating but does not kill the cells.
Figure 1: cells growing in tissue culture petri dish. LEFT panel: Many different cultured
cells, including MEFs, will adhere to the bottom of the dish. Cell culture medium that is pink
contains a dye called phenol red, which aids in determining the pH of the medium; the
medium turns yellow below pH 6.8 and turns bright pink when over pH 8.2. Cells are
incubated at 37ºC in 5% CO2 (to maintain right pH). RIGHT panel: cells adhered to dish at a
density of 70% confluence.
© 2010 Nature Education All rights reserved.
Figure 2: Mouse embryos at different developmental stages
TW 2013
9
Mouse embryos are staged by days after fertilization, which is designated as “E” followed by
the day number. Male and female mice are put in the same cage, and each morning the
females are observed for a hard, vaginal plug that forms after copulation. When a plug is
observed, the stage is considered E0.5 because copulation tends to take place around midnight.
Picture from the Zhang lab (Azusa Inoue and Shinpei Yamaguchi)
Note how fast the embryo’s morphology changes in 24 hours and how fast the embryo is
developing. Birth happens 20 days after fertilization.
TW 2013
10
Drosophila S2 Cell Culture
It is much easier to grow insect cells than mammalian cells. They are much more robust, do
not require CO2 or an incubator (they can grow at room temperature). They are not from
vertebrates and do not require the same strict regulations; Therefore they are more practical to
use in a high school setting.
We will use insect cells the same way scientists use mouse or human cells. We will use these
cells to illustrate basic techniques used in tissue culture, including a proliferation assay and
determining the cytotoxicity (induction of cell death) of some common chemicals. These
types of experiments are often done in early drug trials to determine how a new drug or
chemical compound will affect cells and at what dosage are they killing the cells. The growth
rate of the cells can be determined by counting the live cells under a microscope at 10X
magnification using the trypan blue exclusion assay and a hemacytometer, plating the cells at
a specific density, then counting each day after.
The insect S2 cell line used here was derived from a primary culture of late stage Drosophila
melanogaster embryos by Imogene Schneider in the late 60s-early 70s, as a tool to study the
developmental biology of fruit flies. S2 cells are now mostly used for heterologous protein
expression in insect cells. S2 is a robust cell line that grows without CO2, at room temperature, as a
semi-adherent monolayer or suspension (Figure 2A). The cells are spherical and readily attach to
the bottom of the dish but not so strongly as to require trypsin treatment to detach. The cells are
grown in Schneider Drosophila Medium. Insect medium was originally formulated to mimic the
main physico-chemical properties of the body fluid of the fruit fly by analyzing larval Hemolymph,
and tested to support the growth of excised imaginal disks.
Cell Viability Measurement
To determine the extent to which a compound is cytotoxic, the viability of the treated population of
cells must be assessed. Viability is a measure of the metabolic state of a cell population that is
indicative of the potential for growth. Membrane integrity is a very good indicator of cell viability.
The most common method used in cell culture to check for membrane integrity and to count live
cells is the dye exclusion assay with the vital stain Trypan blue (Figure 3). Cells with intact
membrane will not take up the stain (dye exclusion), whereas leaky membranes or dead or dying
cells will allow the blue dye to get into cells. The white and blue cells are counted under the
microscope to determine the total number of cells and the % of viable cells. If the culture is healthy
at least 95% of the cells should exclude Trypan blue.
TW 2013
11
Figure 3 (A) Brightfield image of Drosophila melanogaster S2 cells; cells are round and semiadherent. (B) Cell suspension stained with Trypan blue, the dark blue cells are dead or dying and
the viable cells have clear centers.
See YouTube for cell counting using the Trypan blue exclusion method:
http://www.youtube.com/watch?v=wPeNK0pRIpA
Induced Pluripotent Stem Cells
Cell replacement therapies: iPS technology or transdifferentiation?
Published: 28 Feb 2011 (http://www.eurostemcell.org/commentanalysis/cell-replacementtherapies-ips-technology-or-transdifferentiation) by Thomas Graf
The ability to convert one cell type into another has caused great excitement in the stem cell field.
Two techniques exist: one reprograms somatic cells into pluripotent stem cells (iPS cells), the other
converts somatic cells directly into other types of specialized cells by transdifferentiation. These
techniques raise high hopes that patient-personalized cell therapies will become a reality in the notso-distant future.
TW 2013
12
A technique developed by Yamanaka in 2006 makes it possible to convert essentially any cell in the
body back into pluripotent stem cells that are almost identical to embryonic stem cells. This is done
by adding a quartet of proteins: the transcription factors Oct4, Sox2, Klf4 and Myc. The resulting
induced pluripotent stem (iPS) cells can be grown and multiplied almost indefinitely without losing
their potential to differentiate into a broad range of cell types. If a clinician wanted to use this
technology to treat a patient with say, Parkinson's disease, she/he would prepare a skin biopsy, grow
skin-derived cells called fibroblasts in the lab, add the Yamanaka combination of four proteins and
wait a couple of months for stable populations or ‘lines’ of induced pluripotent stem cells to be
established. Since iPS cells can proliferate indefinitely, they can be isolated relatively easily and a
small initial population can be used to produce a large number of cells. In our hypothetical
Parkinson’s treatment, the multiplied iPS cells would then be made to differentiate into
dopaminergic neurons, the cell type that is deficient in Parkinson's patients. As a final step the
neurons would be purified and injected back into the patient.
Reprogramming of somatic cells for Induced Pluripotent Stem cell (iPS cell) generation can be
accomplished by a number of technologies. (Picture by THOMAS GRAF)
Comparison of iPS reprogramming and transdifferentiation:
These processes might eventually be applied in the clinic for cell replacement therapies.
An alternative to the iPS procedure is transdifferentiation. This approach uses transcription factors
to convert a given cell type directly into another specialized cell type, without first forcing the cells
to go back to a pluripotent state. Research in the 1980s and 90s showed that fibroblasts can be
converted directly into muscle cells at very high efficiencies using the transcription factor MyoD.
Similarly, scientists found they could use a transcription factor called C/EBPa to turn lymphocytes
TW 2013
13
into macrophages (different types of white blood cell). However, this transdifferentiation approach
has only recently taken off in the stem cell community. One reason for the slow start is that it took
the Yamanaka experiments on iPS to convince many skeptics that cell reprogramming is possible at
all. Another is that for a long time it seemed that direct conversions could only be achieved between
'sister cells', such as between two types of blood cells. The relationship between cell types is
sometimes pictured as a developmental ‘landscape’.
(Graphics by Debbie Maizels)
Landscape of development: The four main germ layers in which cells develop are divided by
‘tectonic plates’. Transitions between cell types are hardest when they cross over tectonic plates.
See also Graf & Enver, 2009 and Waddington, 1957.
Embryonic stem cells and iPS cells sit on a mountain at the very top of the landscape and can
produce cells that fall down into all the different more specialized valleys below. Once the cells are
settled in a particular area, travelling across a ‘tectonic plate’ into a different region to become an
unrelated cell type is a very tough challenge. ‘Sister’ or ‘neighbouring’ cell types can more easily
move over a small hill from one neighbouring valley to the next, if given the right encouragement.
This restriction to 'small jumps' between related cell types kept transdifferentiation firmly within the
realm of basic research studies. Then, in 2010, the barrier was broken. A group of researchers at
Stanford demonstrated that a combination of three neural transcription factors can convert
fibroblasts into functional neurons (Vierbuchen et al., Nature 2010). This study showed that
transcription factors can induce 'large jumps' between distantly related cell types, opening up the
prospect that any desired specialized cell could be generated from essentially any other cell type.
Since then, blood cells have also been generated from fibroblasts (Szabo et. al, Nature 2010),
making it likely that many more such transitions will be reported in the near future.
TW 2013
14
So, which of the two approaches – iPS or transdifferentiation – will make it into the clinic first?
Which will eventually prevail? Because both technologies are patient-specific, there is virtually no
risk of immune rejection. The iPS approach has the advantage that it enables us to obtain large
numbers of cells. Genetic defects can be corrected at the iPS cell stage, meaning that specialized
cells made for the patient no longer have the defect. The disadvantages of iPS are its complexity,
high costs and the length of time required to produce first iPS cells and then the specialized cells
needed for transplantation. There is also a risk that residual iPS cells in the transplant could cause
tumors. The advantages of transdifferentiation are its relative simplicity, lower costs and shorter
times required. However, it is unclear whether it will be possible to generate the large numbers of
specialized cells required for transplantation via the transdifferentiation route. And then there are a
number of concerns common to both techniques. The cells produced for transplantation may not be
fully functional, for example because they retain a 'memory' of their origin. They may not engraft
efficiently or correctly when transferred to the patient, or they may not survive long-term. Finally,
not all degenerative diseases might be amenable to treatment by cell replacement strategies at all,
perhaps even excluding certain disease groups entirely. In conclusion, the verdict is wide open as to
which technique will enter the clinic first, and and for what diseases each approach will be most
effective.
Activation of transcription factor NF-kappaB by TNF-alpha
Tumor necrosis factor (TNF) is involved in the inflammatory response that occurs when an
organism’s immune system is challenged such as in the development of cancer, or upon exposure to
pathogens like bacteria and viruses. When the immune system is challenged, macrophage cells are
activated and secrete TNF-alpha, which stimulate a cellular response from other immune cells. Due
to this property, it is classified as a “cytokine” because it triggers a signaling response in the cells
with which it interacts. When it stimulates other cells of the immune system, those cells then
produce cytokines of their own to participate in a global immune signaling response. It is able to
induce fever, cell death, inflammation and to inhibit tumorigenesis and viral replication.
Dysregulation of TNF-alpha production has been implicated in a variety of human diseases
including Alzheimer's disease, cancer, and inflammatory bowel disease.
In this course, we will explore how TNF-alpha elicits a cellular response by stimulating the NFKappaB signaling pathway (see Figure 4). NF-KappaB (NF-kB) is a transcription factor made of
two protein subunits p65, and p50 [p means protein and the number indicates the molecular weight
of the protein]. NF-kB binds at specific sites on the DNA (called kB sites) and induces/increases the
expression of a number of genes involved in cellular processes such as immune and inflammatory
responses, developmental processes, cellular growth, and apoptosis.
Under normal conditions, NF-kB is present in the cytoplasm of the cell bound to a protein called IKappaB (for inhibitor of NF-kB). I-KappaB keeps NF-kB in the cytoplasm and thus prevent it from
binding to the DNA in the nucleus.
TW 2013
15
Upon exposure to cell stress or stimulation with TNF-alpha, the I-KappaB subunit is
phosphorylated and dissociates from NF-kB. NF-kB possesses a nuclear localization signal (NLS
sequence) which allows it to enter the nucleus, bind to DNA and regulate gene expression.
When I-KappaB is bound to NF-kB it hides the NLS sequence so NF-kB is restricted to the
cytoplasm. If a mutation in either NF-kB or I- kB blocks the interaction between the two proteins
then the activity of NF- kB is no longer regulated and it always goes to the nucleus and activate
gene transcription (including cell growth required in cancer).
Figure 3 TNF-alpha activated NF-KappaB transcription. Adapted to Emily Rosowski
See Slides by Mourad Ali on http://www.slideserve.com/claudia/tumor-necrosis-factor-alpha-tnf-a-acytokine-protein-involved-in-inflammatory-and-immunologic-responses-by-mourad-ali for more on the role
of TNF-alpha
See YouTube clip for activation of NFkappaB: http://www.youtube.com/watch?v=MNVB7K-MDws
DAY 1
MONDAY AFTERNOON
TW 2013
16
Procedure 1: MEF culture
Material:
Mouse embryos, razor blades, 10 cm tissue culture dishes, sterile phosphate buffered saline (PBS),
Trypsin-EDTA, MEF media (Dulbecco’s Modified Eagle Media supplemented with fetal bovine
serum and penicillin/streptomycin antibiotics; DMEM+10% FBS+ Pen/Strep)
Objectives: To learn how to culture primary mouse embryo fibroblasts (MEFs) from mouse
embryos.
**NOTE: Prior to beginning, warm your bottle of DMEM in the 37°C water bath in the tissue
culture room for at least 15 minutes. Remove the bottle from the bath, spray the outside with 70%
Ethanol and wipe dry before setting it into the hood.
Steps 1-3 will be performed by Dr. Aurora Burds Connor in the Koch Institute
1- Embryos are harvested from euthanized pregnant females at E12.5–E13.5, by hysterectomy.
2- The uterine horns containing the embryos are collected in PBS.
3- Embryos are decapitated immediately and washed in PBS several times until most blood is
removed.
Steps 4-13 will be first demonstrated by Aurora Burds Connor.
4- Carefully wipe your bench with 70% ethanol. Make sure you have all necessary material and
media ready on the bench.
5- Obtain one headless mouse embryo and one embryo’s head per team. From this point onwards,
each embryo is processed individually. Keep head and carcass separated to avoid contamination
of carcass with brain tissue.
6- Remove internal organs (anything red or pink colored) with sharp forceps and place onto an
inverted petri dish lid. (The organs will be collected for incineration at the end of the session).
Wash the carcasses extensively in PBS in a 10 cm tissue culture dish.
7- Place the carcass or head on a new 10 cm tissue culture dish in 1 ml of sterile PBS and chop the
embryo with a sterile razor blade into very small pieces (less than 1mm2) until your hand
cramps up.
8- Add 5 ml of Trypsin-EDTA to each dish and transfer dishes to 37°C/5% CO2 incubator for 5
min.
9- During the incubation, take 2 clean 10 cm tissue culture dishes per embryo and add 5 ml sterile
gelatin solution; keep the plates in the incubator until you are done with step 10.
10- Remove the plate from the incubator, and pipette the suspension of trypsin-dissociated cells and
tissue pieces up and down several times with a 10 ml plastic pipette.
*NOTE: Pipette most of the media into a 10ml pipette and with the dish slightly angled, place
the tip of the pipette against the bottom of the dish and expel most but not all of the liquid to
avoid air bubbles.
TW 2013
17
11- Replace the lid to the dish and place the dish back into the incubator; repeat the 5 minute
incubation and pipetting one or two more times (steps 8 and 10) until there are practically no
pieces of tissue left and all the tissue is dissociated into single cells.
12- Remove the 10 cm plates with gelatin solution from the incubator and using a pipet aid and a 10
ml or 25 ml pipette aspirate the gelatin solution completely. You will use these plates for step
13.
13- Transfer the resulting single cell suspension from step 10 to a 50ml conical tube containing 25
ml of DMEM + 10% FCS + Pen/Strep (one embryo per conical), and pipette the solution up and
down 5 times being careful not to draw more than 12 ml of liquid into the pipette, then transfer
12 ml of the suspension per dish onto 2 X 10 cm dishes.
14- Culture the fibroblasts for 2–4 days until the cells reach confluence, meaning the cells have
divided to cover the entire bottom of the dish and there are little visible spaces between the
cells.
15- Observe the progression of the cultures each day and record your results (cellular
morphology/density).
Note: you will need to feed the cells on Wednesday and split them on Thursday.
Procedure 2: Determining cell counts
Each team will obtain a 10 cm tissue culture dish of insect cells and the procedure below will be
carried out on your bench tops in the lab space.
Material: Insect cells: Drosophila melanogaster S2 cells, 10 cm tissue culture dishes, S2 media
(Shields and Sang M3 insect media supplemented with 10% FBS and Pen/Strep), hemacytometer,
Trypan Blue
Objectives: To learn how to count cells for plating at a specific density. NOTE: In contrast to the
MEFs, S2 cells are semi-adherent, and they do not require trypsin for dissociation. Tapping the
plate and pipetting are sufficient to obtain a single cell suspension.
0- Transfer 0.5 ml of Trypan blue into each of four 1.5 ml eppendorf tubes
1- Obtain a 10 cm dish of S2 insect cells. Observe the cells under the inverted microscope at 10X
magnification. The cells should be about 70% confluent. Most cells will be adhering to the
plate and will not move if you gently shake the plate.
2- With the lid on, gently tap the side of the dish with your index finger to begin detaching the
cells from the dish.
3- Remove the lid. Using a 10 ml plastic pipette and pipetaid pipette the media up and down to
resuspend the cells: hold the dish at a 30 degree angle and draw most of the media into the 10
ml pipet, place the tip of the pipette against the bottom of the dish and expel most of the media.
Do not blow bubbles onto the dish (Don’t expel all of the media). Keep an eye on the media
toward the bottom of the dish as you angle it- if you angle it too much the media will spill over
the side.
TW 2013
18
4- Repeat the pipetting 10 times to obtain a suspension of single cells- place the tip of the pipette in
different areas of the dish as you pipette to dissociate all the cells from the bottom of the dish.
5- Observe the dish under the microscope to make sure the cells are completely dissociated from
each other and not clumped or in pairs.
6- Use a p1000 pipetteman and blue filtered tip to immediately remove 0.5ml of the cell
suspension and place it into a 1.5ml eppendorf tube that contains 0.5ml Trypan Blue solution;
cap the tube tightly.
7- Gently invert the cell suspension several times to mix the cell suspension with the Trypan blue
and incubate at room temperature for 5 minutes.
Counting cells with a hemacytometer
See YouTube clip for cell counting using the Trypan blue exclusion method:
http://www.youtube.com/watch?v=wPeNK0pRIpA
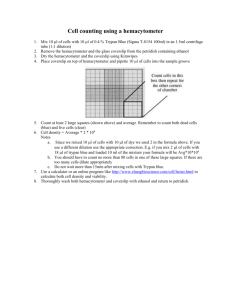
1- Each hemacytometer contains 2 grids for counting cells (delineated by red boxes). Wipe the
hemacytometer surface clean with a Kimwipe, and place the coverslip on top of the grid. If the
surface and/or coverslip has dirt, spray with 70% Ethanol and wipe for better cleaning.
2- Gently invert cell suspension again, and with a p20 pipetteman and p20 filter tip add 10 μl of the
cell suspension into the groove at the top of the grid- make sure solution spreads evenly across
grid and that there are no air bubbles. Then re-cap the tube, invert again, and add 10 µl to the
groove at the bottom of the lower grid. If you get bubbles in either grid, remove coverslip, wipe
clean again, and start over.
3- Count total number of cells in the squares marked 1-4 in the upper grid first; keep separate
counts of live cells (clear centers, luminescent edges) and the dead cells stained with Trypan
Blue. For each square, count cells within the square itself AND on the outer edges of the
square, and exclude cells on the inner edges as per the diagram above. To keep track of the cell
count, you will use a manual cell counter on the desk next to the microscope or a hand clickers.
TW 2013
19
Use one channel for the live cells, and the adjacent channel for the Trypan blue-positive cells,
and combine the totals to get the total number of cells.
4- Determine the average number of cells per square (AVG; total # cells for all 4 chambers ÷ 4).
5- Repeat steps 3 and 4 for the lower grid. Use the average of the 2 AVG values you obtained.
The numbers should be within 10% of each other if they are not exactly the same.
CALCULATIONS
Cells/ml = AVG X 2(dilution factor*) X 104
*Our dilution factor is 2 because we mixed 0.5 ml cell suspension with 0.5 ml Trypan Blue
solution
Total cells = cells/ml X original volume of cell suspension
Cell viability (%) = total # live cells (unstained) ÷ total # cells (unstained and stained) X 100
EXAMPLE
You have a cell suspension of 4mls, and count 150 live cells and 14 Trypan Blue-positive cells
for a total of 164 cells.
AVG= 164 ÷ 4= 41
Cells/ml= 41 X 2 X 104= 8.2 X 105
Total cells= (8.2 X 105) X 5= 3.28 X 106
Cell viability= 150 ÷ 164 X 100= 91.4%
WORKSPACE- CALCULATIONS FOR CELL COUNTS:
TW 2013
20
Count cells from your initial 10cm plate twice to be sure your cell counts are accurate. If your
numbers are not within 10% of each other, count cells a third time. Average the two cell counts to
determine your number of cells/ml.
Count 1:
Count 2:
AVG:
Cells/ml:
Total cells:
TW 2013
21
DAY 2:
Split/plate S2 cells,
Add TNF-alpha /Block overnight
Observe MEFs
MORNING
Procedure 1: Splitting S2 cells for proliferation assay
Material: Drosophila melanogaster S2 cells, tissue culture dishes, S2 media, hemacytometer,
Trypan Blue
Objectives: To plate cells at a specific density to calculate proliferation rates and cell viability
(after treatments) on subsequent days.
NOTE: THIS PROCEDURE WILL BE DONE IN THE TISSUE CULTURE ROOM UNDER
THE HOOD.
1- Upon your arrival at the lab, remove your bottle of S2 media from the refrigerator and warm for
10 minutes in the 37°C water bath in the tissue culture room.
2- During the incubation, spray your tissue culture hood with 70% Ethanol and wipe clean with a
paper towel.
3- Remove the S2 media from the bath, spray it with 70% Ethanol, and wipe it clean with a paper
towel before placing it in the hood.
4- Each team will be given a fresh 10 cm dish of S2 cells.
5- In the tissue culture hood, with the lid on, gently tap the side of the dish to begin dissociating the
cells.
6- Lift the lid to pipette the media up and down: hold the dish at a 30 degree angle and draw most
of the media into a 10ml pipet, place the tip of the pipette against the bottom of the dish and
expel most of the media (if all of it comes out, you will get bubbles). Keep an eye on the media
toward the bottom of the dish as you angle it- if you angle it too much the media will spill over
the side.
7- Repeat the pipetting 10 times to obtain a suspension of single cells- observe the dish under the
microscope to make sure cells are completely dissociated from each other and not clumped or in
pairs.
8- With a p1000 pipetteman and filter tip immediately remove 0.5ml of the cell suspension and
place it into a 1.5ml eppendorf tube that contains 0.5ml Trypan Blue/PBS solution; cap the tube
tightly.
9- Gently invert the cell suspension several times to mix the cell suspension with the Trypan blue
and incubate at room temperature for 5 minutes.
10- Count the LIVE, UNSTAINED CELLS ONLY in this procedure. Resuspend the cells at a
density of 1 X 105 cells/ml (see below for calculation).
WORKSPACE: CALCULATING CELL DENSITY FOR SEEDING PROLIFERATION ASSAY
TW 2013
22
EXAMPLE: You obtain a cell count of 144 live cells in the 4 squares of the hemacytometer.
AVG = 144 ÷ 4 = 36
Cells/ml = 36 X 2 X 104 = 7.2 X 105
DILUTE TO 1 X 105:
ADD 1ml CELL SUSPENSION TO 6.2 ml FRESH MEDIA
Since you need 16 ml, use A 50 ml conical tube and add 3 ml of cell to 18.6 ml of fresh media.
11- Obtain two 6-well plates.
12- Plate 2 ml/well in 4 wells of each of the 6-well plates (you will need a total volume of 16 ml of
diluted cells)
13- Incubate overnight at room temperature
TW 2013
Cells
Cells
Cells
Cells
Cells
Cells
Cells
Cells
23
AFTERNOON
Procedure 2: Treatment of cells with TNF-alpha
Material: Fibroblasts plated in chamber slides, TNF-alpha, PBS, formaldehyde, antibody blocking
solution
Objectives: To observe the cellular localization of NF-KappaB in the presence or absence of TNFalpha
This procedure will take 3 days. You will
Day 1: Treat one chamber of cells with TNF-alpha for 30 minutes, and begin antibody
procedure (Block overnigth)
Day 2: Incubate the cells with anti-p65 primary antibody (this antibody will recognize the p65
subunit of NF-KappaB). Incubate the cells with secondary Ab. This secondary antibody is
labeled with a fluorescent tag and recognizes the primary antibody. Wash and apply coverslips
to the slides.
Day 3: Record results by visualizing p65 localization in the untreated cells compared to the
TNF-alpha treated cells; this will require a fluorescent microscope
PROCEDURE:
NOTE: All washes will be done with a p1000 pipettman and filter tips.
1- Spray the tissue culture hood with 70% Ethanol and wipe clean with a paper towel.
2- Each pair will have a 2-chamber slide containing fibroblasts in the incubator. Remove the slide
from the incubator and place into the hood.
3- Add 10 µl of mouse TNF-α (at a concentration of 20 ng/mL) to the RIGHT chamber.
4- Return slide to the 37°C in the incubator and incubate the cells for 30 min.
5- Bring the slides to your benches. Pipette the media out of the chambers. Never let cells sit
without medium for too long, they will dry out.
6- Wash each chamber once with 0.5 mL of PBS.
7- Remove PBS and fix the cells with 0.5 mL of a solution of 3% Formaldehyde (in PBS) for 20
minutes at room temperature.
8- Remove Formaldehyde solution and wash once with 1mL of PBS.
9- Permeabilize the cells (poke holes in the cell membrane so the antibodies can get in) by adding
0.5 ml of 100% Ethanol at room temperature for 5 minutes.
10- Remove Ethanol and wash X3 with 1mL of PBS, then aspirate the final PBS wash.
11- Block the cells by adding a solution that contains 0.5 ml PBS/ 3% BSA/ 0.2% Triton
X100/ 0.1% Sodium Azide (500 μL per well) overnight at 4°C. This step is required to
reduce background from non-specific binding of the primary antibody that will be used
tomorrow morning.
TW 2013
24
DAY 3:
Observe and feed MEFs
Treat S2 cells with various chemicals (procedure 1)
Continue TNF-alpha experiment (procedure 2)
Transfect S2 cells with Huntingtin gene (procedure 3)
MORNING
**OBSERVE and FEED MEF CULTURES
1- Warm your MEF media in the 37°C water bath for 15-20 minutes.
2- During the incubation, spray the hood glass with 70% Ethanol, lift the sash to the 8 inch mark
and spray the interior of the hood with 70% Ethanol. Wipe surfaces clean with a paper towel.
3- Remove the bottle of MEF media from the bath, spray with 70% Ethanol, wipe clean, and place
in the hood.
4- Remove your MEF plates from the incubator and place them in the hood.
5- Remove the media from the MEF plates with a 10 ml pipette and discard in the liquid waste
container in the hood.
Gently add 10 ml of MEF media to each plate and return the plates to the 37°C incubator
Procedure 1: Cytotoxic treatmentsWe will test 4 different chemicals. Each team will test one chemical. Each treatment will be done
in duplicates or using different concentrations of the same chemical.
This morning we will simply add concentrated doses of ethanol, salt, sugar, or acetic acid to the
bottom wells in the 6-well plates designated for the proliferation assay.
Material: S2 insect cells plated in 6-well chambers from Tuesday, Ethanol, Sodium Chloride,
Glucose, Acetic Acid
Objectives: To determine the effect of common household/dietary items on cell proliferation and
viability by directly comparing proliferation of untreated cells to that of the treated cells. The four
treatments will be compared at the end of the week during the presentations.
1- In the tissue culture room, spray the tissue culture hood with 70% Ethanol and wipe clean with a
paper towel.
2- Observe the wells of your 6-well plates to check that most of your cells are attached- there will
be some floating cells. Then place the plates in the hood.
3- Add the following doses of compounds to your insect cells:
GROUP 1: Ethanol
Stock solution of 17.1M Ethanol (200 proof) is provided. Add 11.75 µl or 47 µl Ethanol/well to
the LOWER 2 wells for each 6-well plate to reach a concentration of 100 mM or 400 mM Ethanol.
Add 11.75 µl or 47 µl of H2O to the TOP 2 wells as control.
TW 2013
25
GROUP 2: Salt (Sodium Chloride, NaCl)
Stock solution of sterile 5M NaCl is provided. Add either 20 µl NaCl/well to the LOWER 2 wells
for each 6-well plate for a final concentration of 50 mM NaCl, or 60 µl for a final concentration of
150 mM. Add the same volume of H2O to the top well for control.
GROUP 3: Acetic Acid
Stock solution of 17.1M Acetic Acid is provided. Add either 8.2 µl Acetic Acid/well to the
LOWER 2 wells for each 6-well plate for a concentration of 50 mM (or 0.3%) Acetic Acid or 30 µl
for a final concentration of 1% acetic acid. Add the same volume of H2O to the top well for control.
GROUP 4: Glucose
Stock solution of 0.5 M Glucose is provided. Add 60 or 180 µl Glucose/well to the LOWER 2
wells for each 6-well plate for a concentration of 10 or 30 mM Glucose.
-
-
-
-
+
+
+
+
A: Untreated control (H2O)
B: Cells treated with EtOH, NaCl, glucose or acetic acid.
Procedure 2: Primary antibody incubation for the TNF-alpha experiment
Material: anti-p65 antibody solution
1- Aspirate blocking solution from each chamber.
2- Add 300 µL of the anti-p65 primary Ab diluted in PBS/3% BSA/0.2% Triton to each chamber
of your slide; incubate at room temperature 1-3 hours (through the seminar and lunch).
Procedure 3: Transfection of S2 cells with wild-type and mutant huntingtin gene.
TW 2013
26
Material: Danielle Manning will demonstrate the transfection procedure, which requires a lipidbased transfection reagent and plasmid DNA that contains the coding sequences of the htt gene. The
lipids in the transfection reagent form a complex with the plasmid DNA that allows the DNA to
effectively enter through the cell membrane. Once inside, the genes present on the plasmid are able
to be transcribed to mRNA and then translated into protein using the cell’s own transcription and
translation machinery. We will use wild-type htt and a mutant called httQ138 that are tagged with
red fluorescent protein (RFP), and can be visualized using a fluorescent microscope.
Objectives: To determine the effect of expressing normal and disease-associated protein in cells.
1- In separate eppendorf tubes, mix 400 ng of each plasmid DNA with 1 µl X-tremeGENE9
transfection reagent in insect media without serum.
2- Incubate at room temperature for 15 min.
3- Add the DNA/X-tremeGENE9 mixture directly to the S2 cells.
4- Observe the cells at 24 and 48 hours after transfection.
AFTERNOON
**OBSERVE THE NEURONAL CELLS provided by today’s speaker (when time permits)
Procedure 2b: Add secondary antibody (TNF-alpha experiment)
Material: Secondary Abs raised in goats against rabbit or mouse IgG; AlexaFluor 488 recognizes
the p65 antibody and AlexaFluor 594 recognizes the tubulin antibody. Hoescht dye stains the
nuclei of cells and will be mixed with the secondary antibodies.
1- Aspirate primary antibody solution and wash X 3 with 1mL of 1X PBS.
2- Add 300 µL of secondary Abs/Hoescht diluted in PBS/3% BSA/0.2% TritonX100; incubate for
1 hour at room temperature in the dark (place slide in a drawer).
3- Aspirate and wash three to five times with 1 mL of 1X PBS leaving the last wash on in dark for
1 hour.
4- Aspirate PBS and remove the chamber portion from the glass slide.
5- Place 3 µL of Vectashield on each chamber.
6- Gently place a glass coverslip on the slide; angle the coverslip and touch it to the bottom of the
slide first, then lay it down slowly to avoid air bubbles.
7- Draw any fluid away from the edges of the coverslip with the corner of a kimwipe.
8- Seal the edges of the coverslip with a thin line of nail polish. Allow to dry and place at 4°C.
Procedure 1b: Count DAY 1 of proliferation assay
1- For each plate, you have 2 wells of untreated cells and 2 wells of treated cells. Count the 2
wells of untreated cells, and 2 wells of treated cells.
2- For each well, label and prepare a separate eppendorf tube with 0.5 ml Trypan blue solution.
TW 2013
27
3- Remove 6-well plates of S2 cells from the cell culture room; cell counts can be performed on
the benchtop, as the cells will be discarded after the counts are complete.
4- With the lid on, gently tap the side of the dish to begin dissociating the cells.
5- Lift the lid to pipette the media up and down in the first well. Be sure not to mix well contents.
6- Repeat the pipetting 10 times.
7- Immediately remove 0.5 ml of the cell suspension with a p1000 and filter tip, and place into a
1.5 ml eppendorf tube that contains 0.5 ml Trypan blue/PBS solution, cap the tube.
8- Gently invert the cell suspension several times and transfer 10 µl to a hemacytometer with a p20
pipetteman and filter tip.
9- Count the cells and record the total number of live, unstained cells as well as the total number of
dead, stained cells. Then clean the hemacytometer and repeat steps 4-7
10- Calculate the cell proliferation and cell viability for each condition as follows:
The assay began with 1 X 105 live cells/well.
To determine how the cells proliferated, calculate the # of cells/ml and the total # of cells and # of
LIVE cells.
AVG = total # live cells ÷ 4
You will determine the AVG for each well
add the AVGs for the 2 untreated wells (-) and ÷ 2; AVG-U
add the AVGs for the 2 treated wells (+) and ÷ 2; AVG-T
Cells/ml = AVG-U (-) X 2 (dilution factor*) X 104
*Our dilution factor is 2 because we mixed 0.5 ml cell suspension with 0.5 ml Trypan Blue
solution
Total cells = cells/ml X original volume of cell suspension
UNTREATED:
Cells/ml = _____ X 2 X 104 = _____
Total cells = _____ X 3 ml = _____
TREATED:
Cells/ml = _____ X 2 X 104 = _____
Total cells = _____ X 3 ml = _____
To determine the number of DEAD cells in each sample, calculate the cells/ml and the total cells for
the STAINED cells.
TW 2013
28
AVG = total # dead cells ÷ 4
You will determine the AVG for each well
add the AVGs for the 2 untreated wells and ÷ 2; AVG-U
add the AVGs for the 2 treated wells and ÷ 2; AVG-T
Cells/ml = AVG-U (or -T) X 2(dilution factor*) X 104
*Our dilution factor is 2 because we mixed 0.5 ml cell suspension with 0.5 ml Trypan Blue
solution
Total cells = cells/ml X original volume of cell suspension
UNTREATED:
Cells/ml = _____ X 2 X 104 = _____
Total cells = _____ X 3 ml = _____
TREATED:
Cells/ml = _____ X 2 X 104 = _____
Total cells = _____ X 3 ml = _____
DETERMINE cell viabilityYou can use the cell counts directly from the hemacytometer tally
Cell viability (%) = total # live cells (unstained) ÷ total # cells (unstained and stained) X 100
UNTREATED:
Cell viability (%) = _______ ÷ _______ X 100
TREATED:
Cell viability (%) = _______ ÷ _______ X 100
DAY 4:
Split MEFs,
Observe p65 localization
Count cells
TW 2013
29
Observe transfected cells
MORNING
**OBSERVE p65 LOCALIZATION
Procedure 1: Split cultured MEFs.
6- Warm your MEF media, PBS, and trypsin aliquot in the 37º C water bath for 15-20 minutesduring this time.
7- During the incubation, spray the hood glass with 70% Ethanol, lift the sash to the 8 inch mark
and spray the interior of the hood with 70% Ethanol. Wipe surfaces clean with a paper towel.
8- Remove the bottles MEF media, PBS, and trypsin from the bath, spray them with 70% Ethanol,
wipe them clean, and place them in the hood.
9- Remove your MEF plates from the incubator and place them in the hood.
10- Remove the media from the MEFs with a 10 ml pipette and discard in the liquid waste container
in the hood.
11- Gently add 5 ml of PBS to each plate to wash away the serum.
12- Remove the PBS and add 1ml 0.5% trypsin to each plate, gently rotate plate to cover all cells
with trypsin.
13- Return to incubator 5-10 min. During this time, label 3 separate fresh 10 cm dishes: 1:2, 1:3,
1:9 so you are prepared for step 10.
14- Add 8 ml media to plates and pipette up and down several times to obtain single cell suspension
(this will give you 9 ml total).
15- Split at the following ratios into 3 separate plates: 1:2 (4.5 ml into plate 1), 1:3 (3 ml into plate
2), 1:9 (1ml into plate 3).
AFTERNOON:
**OBSERVE HEART CELLS DERIVED FROM STEM CELLS provided by today’s speaker
**OBSERVE THE S2 CELLS TRANSFECTED WITH WILD-TYPE AND MUTANT htt gene
Procedure 1: Count cells, complete proliferation assay
1- For each plate, you have 2 wells of untreated cells (-) and 2 wells of treated cells (+). Count the
2 wells of untreated cells, and 2 wells of treated cells. For each well, label and prepare a
separate eppendorf tube with 0.5 ml Trypan blue solution.
2- Remove 6-well plates of S2 cells from the cell culture room; cell counts can be performed on
the benchtop, as the cells will be discarded after the counts are complete.
3- With the lid on, gently tap the side of the dish to begin dissociating the cells.
4- Lift the lid to pipette the media up and down in the first well. Be sure not to mix well contents.
5- Repeat the pipetting 10 times.
TW 2013
30
6- Immediately remove 0.5 ml of the cell suspension with a p1000 and filter tip, and place into a
1.5 ml eppendorf tube that contains 0.5 ml Trypan blue/PBS solution, cap the tube.
7- Gently invert the cell suspension several times and transfer 10 µl to a hemacytometer with a p20
pipetteman and filter tip.
8- Count the cells and record the total number of live, unstained cells as well as the total number of
dead, stained cells. Then clean the hemacytometer and repeat steps 4-7
9- Calculate the cell proliferation and cell viability for each condition as follows:
The assay began with 1 X 105 live cells/well.
To determine how the cells proliferated, calculate the cells/ml and the total cells for the LIVE
cells.
AVG = total # live cells ÷ 4
You will determine the AVG for each well
-add the AVGs for the 2 untreated wells and ÷ 2; AVG-U
-add the AVGs for the 2 treated wells and ÷ 2; AVG-T
Cells/ml= AVG-U (or -T) X 2(dilution factor*) X 104
*Our dilution factor is 2 because we mixed 0.5ml cell suspension with 0.5 ml Trypan Blue
solution
Total cells= cells/ml X original volume of cell suspension
UNTREATED:
Cells/ml = _____ X 2 X 104 = _____
Total cells = _____ X 3 ml = _____
TREATED:
Cells/ml= _____ X 2 X 104= _____
Total cells= _____ X 3 ml= _____
To determine the number of DEAD cells in each sample, calculate the cells/ml and the total cells for
the STAINED cells.
AVG= total # dead cells ÷ 4
You will determine the AVG for each well
-add the AVGs for the 2 untreated wells and ÷ 2; AVG-U
-add the AVGs for the 2 treated wells and ÷ 2; AVG-T
TW 2013
31
Cells/ml = AVG-U (or -T) X 2(dilution factor*) X 104
*Our dilution factor is 2 because we mixed 0.5 ml cell suspension with 0.5 ml Trypan Blue
solution
Total cells= cells/ml X original volume of cell suspension
UNTREATED:
Cells/ml = _____ X 2 X 104 = _____
Total cells = _____ X 3 ml = _____
TREATED:
Cells/ml = _____ X 2 X 104 = _____
Total cells = _____ X 3 ml = _____
DETERMINE Cell viabilityyou can use the cell counts directly from the hemacytometer tally
Cell viability (%) = total # live cells (unstained) ÷ total # cells (unstained and stained) X 100
UNTREATED:
Cell viability (%) = _______ ÷ _______ X 100
TREATED:
Cell viability (%) = _______ ÷ _______ X 100
RESULTS OF PROLIFERATION ASSAY:
DAY 5:
OBSERVATIONS:
TW 2013
32
Passaged MEFs- did the plating density affect the cells?
Transfected S2 cells
Clean up
Appendix 1: Insect media recipe and tissue culture supplies (including sterile solutions for cytotoxicity
experiments)
S2 insect media RECIPE
Shields and Sang M3 insect media
TW 2013
445 ml
33
FBS, heat inactivated
Penicillin/Streptomycin
Total
50 ml
5 ml
500 ml
Product
Catalog number
Quantity
10cm culture dish
25382-166
1 case (200)
6-well plates
62406-161
2 cases (50, each)
5ml pipettes
89130-896
1 case (200)
10ml pipettes
89130-898
1 case (200)
25ml pipettes
89130-900
1 case (200)
Trypan blue
97063-702
1 bottle (100ml)
VWR Scientific (https://us.vwr.com/)
SIGMA-ALDRICH (http://www.sigmaaldrich.com)
Shields and Sang M3 insect media
S3652
500ml
FBS, heat inactivated
45000-736
500ml
Acetic Acid
A6283
100ml
Glucose Solution
G8644
100ml
INVITROGEN (http://www.invitrogen.com)
Pennicilin/Streptomycin Antibiotic
15070063
100ml
5M Sodium Chloride
AM9760G
100ml
catalog number
quantity
25382-166
62406-161
1 case (200)
2 cases (50, each)
product
VWR
10cm culture dish
6-well plates
TW 2013
34
2-chamber slides
razor blades
FBS, heat inactivated
PBS
5ml pipettes
10ml pipettes
25ml pipettes
100% EtOH
Trypan blue
Formaldehyde
Triton
62407-292
55411-055
45000-736
45000-434
89130-896
89130-898
89130-900
89085-244
97063-702
MK501602
AAA16046-AE
1 case (16)
1 box (100)
500ml
1 case (6X500ml)
1 case (200)
1 case (200)
1 case (200)
4L bottle
1 bottle (100ml)
1 bottle (500ml)
1 bottle (100ml)
SIGMA
Shields and Sang M3 media
Acetic Acid
Glucose Solution
Gelatin
BSA
S3652
A6283
G8644
G1393
A9647
500ml
100ml
100ml
20ml
smallest size
15070063
AM9760G
25200056
100ml
100ml
100ml
11995073
A-11008
A-11005
H3570
10X 500ml
1
1
1
Santa Cruz
p65
tubulin
Sc-8008
Sc-5546
1
1
MEF media RECIPE
DMEM
FBS, heat inactivated
Pennicilin/Streptomycin Antibiotic
total
445 ml
50 ml
5 ml
500 ml
INVITROGEN
Pen Strep
5M NaCl
Trypsin
DMEM (glucose, L-glut,
NaPyruvate)
AlexaFluor 488, goat anti-rabbit
AlexaFluor 594, goat anti-mouse
Hoechst
TW 2013
35