Sagnelli et al., 2015 – Supplemental data @ Journal of Neurology

Sagnelli et al.
, 2015 – Supplemental data @ Journal of Neurology – Page 1 out of 4
Early-onset progressive spastic paraplegia caused by a novel TUBB4A mutation: brain MRI and
FDG-PET findings.
Anna Sagnelli, Stefania Magri, Laura Farina, Luisa Chiapparini, Giorgio Marotta, Davide Tonduti,
Monica Consonni, Graziana Scigliuolo, Riccardo Benti, Davide Pareyson, Franco Taroni, Ettore
Salsano, Daniela Di Bella.
Corresponding author
: Dr. Ettore Salsano, Fondazione IRCCS Istituto Neurologico “C. Besta”, via
Celoria 11, 20133 Milano, Italy. Phone: +39 02 2394.2388; Fax: +39 02 2394.2293; E-mail: ettore.salsano@istituto-besta.it
.
Sagnelli et al.
, 2015 – Supplemental data @ Journal of Neurology – Page 2 out of 4
GENETIC STUDIES
TUBB4A encodes tubulin β-4A, one of the members of the highly conserved beta-tubulins; t ogether with α-tubulin, β-tubulin is the principal constituent of microtubules . Heterozygous mutations observed in patients with H-ABC syndrome occur in all TUBB4A protein domains and probably alter microtubule dynamics or stability, by mean of dominant negative, toxic effects rather than a loss of function mechanism.
The proband was screened for TUBB4A mutations by using an amplicon-based customized gene panel
(Illumina TrueSeq Custom Amplicon, TSCA) on an Illumina MiSeq apparatus, covering 8 known genes ( AIMP1 , FAM126A , GJC2 , HSPD1 , PLP1 , POLR3A , POLR3B , TUBB4A ) associated with hypomyelinating leukodystrophy. Reads were mapped against the hg19 standard reference genome and analyzed for TUBB4A (GenBank accession No. NM_006087.2) variants. A novel heterozygous A-to-T transversion at nucleotide 1064 (c.1064A>T) was identified in exon 4; this nucleotide change leads to an aspartate-to-valine change at amino acid position 355 (p.Asp355Val) of the TUBB4A protein. This variant is not present in available genetic databases (1000 Genomes, Exome Variant Server, and dbSNP) [1-3].
The p.Asp355Val mutation affects a highly conserved residue, and is predicted to be deleterious by in silico analysis with three different algorithms (PolyPhen-2 [4], SIFT (Sorting Intolerant From Tolerant)
[5], and MutationTaster [6]). Presence of the mutation identified was verified by standard Sanger sequencing. Parental DNA was investigated by Sanger sequencing of the amplicon containing the index patient’s mutation.
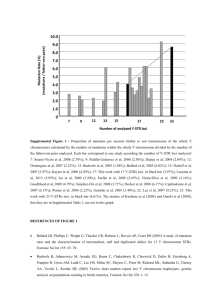
The c.1064A>T variant identified in the patient was not identified in either parent and in his sister, suggesting that it is a de novo mutation (see the family tree below;
–ve, negative; +ve, positive).
Sagnelli et al.
, 2015 – Supplemental data @ Journal of Neurology – Page 3 out of 4
Sanger sequence electropherograms of the
TUBB4A c.1064A>T (p.Asp355Val) mutation in the patient and his parents. Mutation nomenclature is based on the TUBB4A transcript reference sequence (GenBank accession No.
NM_006087.2). Nucleotides are numbered according to the cDNA sequence, with nucleotide +1 corresponding to the A of the ATG translation initiation codon in the reference sequence.
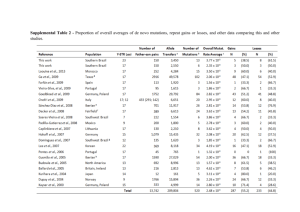
Pathogenicity prediction for the p.Asp355Val mutation
TUBB4A Effect Conservation PolyPhen-2
(0, benign;
1, damaging)
SIFT
(<0.05, deleterious; >0.5, tolerated)
0.00
MutationTaster c.1064A>T
(p.Asp355Val)
Amino acid change
From Xenopus to
Homo
0.990 Disease causing
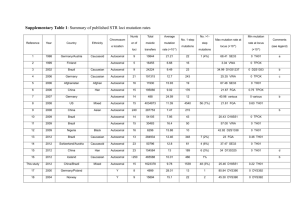
Conservation of the TUBB4A p.D355 residue.
Orthologous protein alignment shows that aspartic acid (D) 355 is highly conserved during evolution (from X. levis to human). gi|574584803|Homo AACDPRHGRYLTVAAVFRGRMSMKEVDEQMLSVQSKNSSYFVEWIPNNVKTAVC D IPPRG360 gi|525343320|Pan AACDPRHGRYLTVAAVFRGRMSMKEVDEQMLSVQSKNSSYFVEWIPNNVKTAVC D IPPRG360 gi|12851187|Mus AACDPRHGRYLTVAAVFRGRMSMKEVDEQMLSVQSKNSSYFVEWIPNNVKTAVC D IPPRG360 gi|47940377|Danio AACDPRHGRYLTVAAVFRGRMSMKEVDEQMLNVQNKNSSYFVEWIPNNVKTAVC D IPPRG360 gi|148224397|Xenopus AACDPRHGRYLTVAAVFRGRMSMKEVDEQMLNIQNKNSSYFVEWIPNNVKTAVC D IPPRG360
Sagnelli et al.
, 2015 – Supplemental data @ Journal of Neurology – Page 4 out of 4
REFERENCES
1.
The 1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA,
Durbin RM et al (2012) An integrated map of genetic variation from 1,092 human genomes.
Nature 491:56-65 ( http://www.1000genomes.org
).
2.
Exome variant server (EVS) of the National Heart, Lung, and Blood Institute GO Exome
Sequencing Project ( http://evs.gs.washington.edu/EVS/ ).
3.
dbSNP [Build ID: 137] ( http://www.ncbi.nlm.nih.gov/SNP/ ).
4.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249
( http://genetics.bwh.harvard.edu/pph2/ ).
5.
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protocols 4:1073–1081
( http://sift.jcvi.org/ ).
6.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D (2010) Mutation Taster evaluates disease - causing potential of sequence alterations. Nat Methods 7:575–576
( http://www.mutationtaster.org/ ).