pmic12047-sup-0001-SuppMat
advertisement

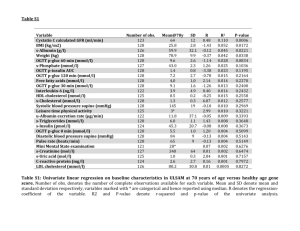
Supporting Information Inhibitor-based affinity probes for the investigation of JAK signaling pathways Michael Höfener1, Fiona Pachl2, Bernhard Kuster2,3, Norbert Sewald1* 1 Organic and Bioorganic Chemistry, Department of Chemistry, Bielefeld University, Universitätsstraße 25, 33615 Bielefeld, Germany 2 Chair of Proteomics and Bioanalytics, Center of Life and Food Sciences Weihenstephan, Technische Universität München, Freising, Germany 3 Center for Integrated Protein Science Munich, Freising, Germany *Corresponding author: Prof. Dr. Norbert Sewald, norbert.sewald@uni-bielefeld.de, fax: int+49 (0)521 106 156963 Materials and methods for chemical synthesis Chemicals and Solvents: Solvents were usually used as purchased in p.a. quality. Solvents of lower quailty were purified by distillation. If necessary, methylene chloride was dried over calcium hydride. Toluene and tetrahydrofuran were dried over sodium. Acetonitrile for analytical and preparative HPLC was purchased in Lichrosolv quality from Merck. Water for this purpose was obtained from a Millipore MilliQ-Ultrapure water system. Chemicals were purchased at Acros, Sigma Aldrich, VWR and Alfa Aesar and were used without further treatment. Silica gel (0.04-0.063 mm) Macherey-Nagel was used for flash chromatography. NMR-spectroscopy: 1H-NMR and 1H,1H-COSY experiments were recorded using a Bruker DRX 500 instrument (1H-NMR: 500.1 MHz), 13C-NMR, HSQC and HMBC experiments were recorded using a Bruker Avance 600 instrument (1H-NMR: 600.1 MHz , 13C-NMR: 150.9 MHz). The recorded data was processed using TopSpin v 2.1, the spectra were evaluated using MestReNova software 6.0.2. Deuterated chloroform was usually used as solvent for NMR measurements with tetramethylsilane 1 (TMS) as internal standard. Other deuterated solvents were referenced to the solvent peak. Structures of unpublished molecules were assigned with the help of 1H,1H-COSY, HSQC and HMBC NMR experiments. Mass-spectrometry: ESI-MS: ESI/APCI mass spectra were recorded using an Esquire 3000 ion trap mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany) equipped with a standard ESI/APCI source. Samples were introduced by direct infusion with a syringe pump. Nitrogen served both as the nebulizer gas and the dry gas. Nitrogen was generated by a Bruker nitrogen generator NGM 11. Helium served as cooling gas for the ion trap and collision gas for MSn experiments. The spectra were recorded with the Bruker Daltonik esquireNT 5.2 esquireControl software by the accumulation and averaging of several single spectra. DataAnalysis™ software 3.4 was used for processing the spectra. MALDI-MS: MALDI experiments were performed using a Fourier Transform Ion Cyclotron Resonance (FT-ICR) mass spectrometer APEX III (Bruker Daltonik GmbH, Bremen, Germany) equipped with a 7.0 T, 160 mm bore superconducting magnet (Bruker Analytik GmbH – Magnetics, Karlsruhe, Germany), infinity cell, and interfaced to an external MALDI ion source. Nitrogen served both as the nebulizer gas and the dry gas for ESI. Nitrogen was generated by a Bruker nitrogen generator NGM 11. Argon served as cooling gas in the infinity cell and collision gas for MSn experiments. Scan accumulation and fourier transformation were performed with XMASS NT (7.08) on a PC Workstation, for further data processing DataAnalysis™ 3.4 was used. LC-MS: LC-MS analysis was accomplished using a Waters Alliance HT equipped with a Waters Symmetry 3.5 µm column (C8, 100 x 2.1 mm, eluent A H2O/HCOOH = 100:0.1, eluent B CH3CN/HCOOH = 100:0.1, flowrate 0.4 mL⋅min-1 using a gradient from 5-95% B over 10 minutes) coupled with a Waters micromass ZQ2000 ESI-MS. Analytical Reversed Phase-High Performance Liquid Chromatography (RP-HPLC) Thermo Separation Products System: Surveyor Autosampler Plus, Surveyor LC Pump Plus, Surveyor PDA Plus Detector (190 -800 nm, continous); column: ET 125/4 Nucleosil 100-5 C18 PPN; software: Chromquest 5.0; eluent A: 5 % H2O, 95 % ACN, 0.05 % TFA; eluent B: 95 % H2O, 5 % ACN, 0.05 % TFA. Supplementary table 1: Analytical HPLC method, λ = 192-480 nm. time [min] eluent A eluent B flow [mL min-1] 0 0 100 0.75 1 0 100 0.75 4 100 0 0.75 5 0 100 0.75 Preparative Reversed Phase - High Performance Liquid Chromatography (RP-HPLC) Hitachi MERCK LaChrom system: LC 7150 pump; UV-Vis L 7420 detector; precolumn: Vydac highperformance guard column (C18); column: Phenomenex Jupiter 10 µ 300 Å (C18; 250 x 21.2 mm); eluent A: 5 % H2O, 95 % ACN, 0.05 % TFA; eluent B: 95 % H2O, 5 % ACN, 0.05 % TFA. 2 Supplementary table 2: Preparative HPLC method, λ = 254 nm. time [min] eluent A eluent B flow [mL min-1] 0 0 100 10 5 0 100 10 40 100 0 10 50 0 100 10 Microwave assisted synthesis: Reactions were performed with a CEM Discover instrument, CEM Matthews Inc. (USA), max. 300 W, max. 2455 MHz. The reaction containers were sealed during the heating progress that was run as stated in each corresponding synthetic procedure. Synthesis of probe 1b: Supplementary Figure 1: Overview of the synthesis of probe 1b. tert-Butyl 2-(2-(2-aminoethoxy)ethoxy)ethylcarbamate (1) A mixture of Boc2O (2.9 g, 14 mmol) in THF (30 mL) was added dropwise over a period of 5 h to a solution of 2-(2-(2aminoethoxy)ethoxy)ethanamine (14.5 g, 98 mmol) in THF (30 mL). The resulting suspension was stirred overnight at rt prior evaporation of the solvent. The residue was dissolved in water (50 mL) and extracted with DCM (3x20 mL). The organic phases were washed with water (10 mL) dried over MgSO4 and the solvent evaporated. The crude product was used without purification for the next step. Formula: C11H24N2O4. Yield: 3.3 g (13.3 mmol, 95%). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 5.16 (s, 1H, NH), 3.62 (s, 4H, 2xCH2), 3.54 (m, 4H, 2xCH2), 3.32 (m, 2H, CH2), 2.90 (t, 2H, J = 5.1 Hz, CH2), 1.44 (s, 9H, tBu). tert-Butyl 2-(2-(2-(cyclohexylmethylamino)ethoxy)ethoxy)ethylcarbamate (2) Cyclohexylmethyl bromide (18 µL, 27.0 mg, 0.15 mmol) was dissolved in dry DCM (1 mL) and added dropwise over 4 h to a solution of amine 1 (47.2 mg, 0.19 mmol) and TEA (29.1 µL, 21.0 mg, 0.21 mmol) in DCM (1 mL) at rt. After stirring overnight at rt the solvent was evaporated to give the crude product that was purified via flash chromatography (eluent: DCM/MeOH, 9.5:0.5 +1% TEA). Formula: C18H36N2O4. Yield: 37 mg (0.11 mmol, 73%). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 5.18 (s, 1H, NH), 3.61 (m, 6H, 3 3xCH2), 3.53 (t, 2H, J = 5.3 Hz, CH2), 3.31 (m, 2H, CH2), 2.82 (t, 2H, J = 5.2 Hz, CH2), 2.40 (m, 2H, CH2), 1.60-187 (m, 5H, 2xCH2 1xCH), 1.43 (s, 9H, tBu), 1.10-1.20 (m, 6H, 3xCH2). tert-Butyl2-(2-(2-((cyclohexylmethyl)(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)ethoxy)ethoxy)ethyl carbamate (3) A microwave tube was charged with amine 2 (37 mg, 0.11 mmol) 6-chloro-7deazapurine (17 mg, 0.11 mmol) and TEA (21 mL, 15 mg, 0.15 mmol). The tube was sealed and heated for 120 min to 150 °C (max 40 W) in the microwave. The crude product was purified using flash chromatography (eluent: EE/DCM, 3:1) to give the title compound as an off-white solid. Formula: C42H54F2N6O6. Yield: 33 mg (0.072 mmol, 65%). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 11.84 (s, 1H, ArNH), 8.27 (s, 1H, ArH), 7.05 (d, 1H, J = 3.4 Hz, ArH), 6.42 (d, 1H, J = 3.4 Hz, ArH), 5.08 (m, 1H, NH), 3.95 (t, 2H, J = 6.2 Hz, PEG-CH2), 3.75 (t, 2H, J = 6.2 Hz, PEG-CH2), 3.64 (d, 2H, J = 7.4 Hz, CH-CH2), 3.57-3.62 (m, 4H, 2xPEG-CH2), 3.52 (t, 2H, J = 4.7 Hz, PEG-CH2), 3.29 (m, 2H, PEG-CH2), 1.87 (m, 1H, CH2-CH), 1.61-1.79 (m, 6H, Chex-CH2), 1.43 (s, 9H, tBu), 1.14-1.20 (m, 2H, Chex-CH2), 0.98-1.06 (m, 2H, Chex-CH2). 13C-NMR (150 MHz, CDCl3) : δ [ppm] = 156.7 (ArC), 156.0 (Boc-C=O), 151.7 (ArC), 150.1 (ArC-H), 120.1 (ArC-H), 102.1 (ArC), 101.7 (ArC-H), 79.1 (Boc-CCH3), 70.6 (PEG-CH2), 70.3 (PEGCH2),69.1 (PEG-CH2), 56.3 (CH2-CH), 50.2 (PEG-CH2), 40.3 (PEG-CH2), 37.5 (CH), 30.8 (PEG-CH2), 28.4 (3xBoc-CH3), 26.4 (2xCH2), 25.9 (2xCH2), 14.1 (CH2). N-(2-(2-(2-Aminoethoxy)ethoxy)ethyl)-N-(cyclohexylmethyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (4) The carbamate 3 (33 mg, 0.072 mmol) was dissolved in 1 mL of a mixture of TFA, water and TIS (95:2.5:2.5) and stirred for 1 h at rt. The reaction mixture was coevaporated with isopropanol (2x10 mL) and dried in vacuum to give the title compound as its corresponding ditrifluoro acetate. Formula: C42H54F2N6O6. Yield: 33 mg (0.072 mol, quant.). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 11.14 (s, 1H, ArNH), 8.29 (s, 1H, ArH), 7.81 (m, 3H, NH3+), 7.20 (m, 1H, ArH), 6.54 (m, 1H, ArH), 4.01 (m, 2H, PEG-CH2), 3.55-381 (m, 10H,2xCH2CH+8x PEG-CH2), 3.15 (m, 2H, PEG-CH2), 1.87 (m, 1H, CH2-CH), 1.61-1.79 (m, 6H, Chex-CH2), 1.14-1.20 (m, 2H, Chex-CH2), 0.98-1.06 (m, 2H, Chex-CH2). 13C-NMR (150 MHz, CDCl3) : δ [ppm] = 142.3 (ArC), 123.2 (ArC), 116.7 (ArC-H), 114.8 (ArC-H), 103.6 (ArC), 102.1 (ArC-H), 70.2 (PEG-CH2), 68.2 (PEG-CH2),66.3 (PEG-CH2), 39.7 (CH2-CH), 30.5 (PEG-CH2), 26.1 (PEGCH2), 25.6 (CH), 17.6 (PEG-CH2), 17.2 (2xCH2), 12.2 (2xCH2), 11.6 (CH2). Exact mass (ESI): mz1 = 362.25490 [M+H]+, calc.: 362.25505. 4 Synthesis of probe 2b: Supplementary Figure 2: Overview of the synthesis of probe 2b. 2-Bromo-6-fluoroaniline (5) (Bioorganic & Medicinal Chemistry Letters 2010, 20, 2609–2613) 2Bromo-6-fluorobenzoic acid (1 g, 5 mmol) was dissolved in 95% sulfuric acid (12 mL) and heated to 75 °C for 30 min. The reaction mixture was allowed to cool to rt prior to addition of Sodium azide (429 mg, 6.6 mmol) in three portions over 30 min. After stirring over night at rt the reaction mixture was cooled to 0-4 °C and neutralized with 25% Ammonia solution. The resulting suspension was extracted with EE (5x50 mL) and the organic phases washed with brine and dried over Na2So4. Evaporation of the solvent gave the crude product as viscous oil which was used as obtained in the next step. Formula: C6H5BrFN. Yield: 918 mg (4.8 mmol, 95%). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 7.19 (d, 1H, J = 8.1 Hz), 6.95 (ddd, 1H, J = 10.6, 8.2, 1.3 Hz), 6.57 (td, 1H, J = 8.2, 5.5 Hz). 1-Bromo-3-fluoro-2-nitrobenzene (6) (Bioorganic & Medicinal Chemistry Letters 2010, 20, 2609– 2613) 2-Bromo-6-fluoroaniline (5) (950 mg, 5.0 mmol) as obtained in the previous step was dissolved in DCE (25 mL) and treated with meta-chloroperbenzoic acid (3.9 g, 22.6 mmol). The reaction mixture was heated to 70 °C overnight. After cooling to rt EE (25 mL) was added and the organic phase was washed with 0.1M NaOH solution till complete removal of the perbenzoic acid. After washing with brine and drying over Na2SO4 the solvent was evaporated to give the crude product as a yellow viscous oil that was used without further purification in the next step. Formula: C6H3BrFNO2. Yield: 1.100 g (5.00 mmol, quantitative) 1H-NMR (500 MHz, CDCl3): δ [ppm] = 7.49 (dt, 1H, J = 8.2, 1.3 Hz), 7.38 (td, 1H, J = 8.3, 5.5 Hz), 7.25 (td, 1H, J = 8.7, 1.0 Hz). 5 Ethyl 2-([3-bromo-2-nitrophenyl]amino)acetate (7) (Bioorganic & Medicinal Chemistry Letters 2010, 20, 2609–2613) 1-Bromo-3-fluoro-2-nitrobenzene (6) (1.00 g, 4.5 mmol) as obtained in the previous step was mixed with DIPEA (3.04 mL, 2.30 g, 18 mmol) in DMF (5 mL) and heated to 80 °C. Glycine ethyl ester hydrochloride (1.88 g, 13.5 mmol) was added in three portions during 5 h. After allowing the reaction mixture to cool down to rt the volatile components were evaporated. The crude product was purified using flash chromatography (eluent: PE/EE 9:1) to give an orange crystalline solid. Formula: C10H11BrN2O4. Yield: 950 mg (3.1 mmol, 69%). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 7.16 (t, 1H, J = 8.2 Hz), 7.02 (dd, 1H, J = 7.9, 1.1 Hz), 6.61 (m, 1H), 6.09 (t, 1H, J = 5.3 Hz), 4.27 (q, 2H, J = 7.2 Hz), 3.95 (d, 2H, J = 5.0 Hz), 1.30 (t, 3H, J = 7.1 Hz). 8-Bromoquinoxalinone (8) (Bioorganic & Medicinal Chemistry Letters 2010, 20, 2609–2613) Ethyl 2([3-bromo-2-nitrophenyl]amino)acetate (7) (112 mg, 0.37 mmol) was dissolved in EtOH/H2O 95:5 (2 mL) and treated with Fe powder (62 mg, 111 mmol) and CaCl2 (41 mg, 0.37 mmol). The reaction mixture was heated to reflux for 3 h. After cooling to rt the resulting suspension was filtrated and the filtrate evaporated to dryness. The residue was dissolved in EtOH/THF 1:1 (2 mL) and treated with KOtBu (166 mg, 1.4 mmol) Oxygen was bubbled through the solvent for 1 h and the reaction mixture stirred overnight under oxygen atmosphere at rt. After neutralizing the resulting suspension with 1 M HCl-solution the reaction mixture was evaporated to dryness resolved in DCM (10 ml) and washed with brine (2x5 mL). The organic phase was dried over Na2SO4 and the solvent removed under reduced pressure to give the crude product as a light brown solid that was purified using preparative RP-HPLC. Formula: C8H6BrN2O. Yield: 49 mg (0.22 mmol, 60% over two steps). 1H-NMR (500 MHz, (CD3)2SO): δ [ppm] = 11.66 (s, 1H), 8.21 (s, 1H), 7.85 (d, 1H, J = 7.2 Hz), 7.80 (d, 1H, J = 7.6 Hz), 7.37 (m, 1H). 8-Bromo-2-chloroquinoxaline (9) (Bioorganic & Medicinal Chemistry Letters 2010, 20, 2609–2613) 8Bromoquinoxalinone (8) (49 mg, 0.22 mmol) was dissolved in POCl3 (1.5 mL) and heated for 3 h to 80 °C. The reaction mixture was allowed to cool to rt and unreacted POCl3 was removed under reduced pressure. Water (15 mL) and DCM (15 mL) were added to the residue at 4 °C and the phases were separated. The organic phase was washed with sat. NaHCO3-solution, brine (10 mL) and dried over Na2SO4.The solvent was evaporated to give the crude product that was used as obtained for the next step. Formula: C8H4BrClN2. Yield: 44 mg (0.18 mmol, 82%). 1H-NMR (500 MHz, (CD3)2SO): δ [ppm] = 9.08 (s, 1H), 8.30 (d, 1H, J = 7.5 Hz), 8.19 (d, 1H, J = 8.3 Hz), 7.84 (dd, 2H, J = 8.3, 7.5 Hz). 4-(4-Bromo-2,6-difluorophenyl)morpholine (10) (WO 2008/148867 A2, PCT/EP2008057058) (4Bromo-2,6-difluorophenyl)methanol (179 mg, 0.80 mmol) was dissolved in dry DCM (8 mL) treated with phosphorus tribromide (380 µL, 1.08 g, 4.00 mmol) and stirred overnight at rt. Cold methanol (5 mL) was added and the organic phase was washed with sat. NaHCO3-solution (2x2 mL) before evaporation of the solvent. The residue was dissolved in dry THF (8 mL) and treated with morpholine (694 µL, 697 mg, 8 mmol) and TEA (223 µL, 162 mg, 1.6 mmol) before stirring for 3 h at rt. After adding DCM (20 mL) the reaction mixture was washed with 1M NaOH-solution (2x5 mL) dried over Na2SO4 and the solvent evaporated to give the crude product that was used as obtained for the next step. Formula: C11H12BrF2NO. Yield: 165 mg (0.57 mmol, 70%). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 7.09 (m, 2H, ArH), 3.68 (m, 4H, 2xO-CH2), 3.62 (s, 2H, Ar-CH2), 2.48 (m, 4H, 2xN-CH2). 4-(4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolanyl)2,6-difluorophenyl)morpholine (11) (Angew. Chem. 2010, 122, 7320-7322) 4-(4-Bromo2,6-difluorophenyl)morpholine (10) (106 mg, 0.36 mmol), bispinacolatodiboron (120 mg, 0.47 mmol) and KOAc (71.0 mg, 0.72 mmol) were dissolved in DMF 6 (1 mL). After 5 cycles of freeze-pump-thaw PdCl2(dppf) (29 mg, 0.04 mmol) was added prior to heating the reaction mixture for 60 min at 80 °C in the microwave (max. 20 W). After allowing to cool to rt EE (15 mL) was added and the organic phase washed with water (1x5 mL) sat. NaHCO3-solution (1x5 mL) and brine (1x5 mL). After drying over Na2SO4 and evaporation of the solvent the crude product was purified using flash chromatography (eluent: EE + 1% TEA) to give the desired product as viscous oil. Formula: C17H24BF2NO3. Yield: 87 mg (0.26 mmol, 72%). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 7.30 (m, 2H, ArH), 3.65-3.75 (m, 6H, 2xAr-CH2 + 4xO-CH2), 2.51 (m, 4H, N-CH2), 1.34 (s, 12H, 4xCH3). 2-(2-(2-(2-Azidoethoxy)ethoxy)ethoxy)ethanol (12) Tetraethyleneglycol (54.3 g, 280 mmol) and TEA (30.0 mL, 21.8 g, 215 mmol) were mixed in dry THF (200 mL)and cooled down to 0 °C prior to adding methane sulfonyl chloride (7.9 mL, 11.7 g, 102 mmol) over 45 min. After allowing to warm up to rt the resulting mixture was stirred at rt overnight. The solvent was evaporated and the residue was treated with ethanol (200 mL) and NaN3 (13.0 g, 200 mmol). The reaction mixture was heated to reflux overnight. After evaporation of the solvent the residue was extracted with Et2O (5x100 mL) the residue treated with H2O (200 mL) and extracted with Et2O (3x30 mL). The combined organic layers were washed with brine (2x100 mL) dried over MgSO4 and evaporated to give the crude product that was purified using flash chromatography (eluent: DCM/MeOH 49:1). Formula: C8H17N3O4. Yield: 4.5 g (20 mol, 20% over two steps). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 3.71 (m, 2H, O-CH2), 3.62-3.68 (m, 10H, 5xO-CH2), 3.59 (m, 2H, O-CH2), 3.38 (m, 2H, N-CH2), 2.62 (s, 1H, OH). 13C-NMR (150 MHz, CDCl3) : δ [ppm] = 72.5 (HO-CH2), 70.8 (O-CH2), 70.7 (O-CH2), 70.6 (O-CH2), 70.4 (O-CH2), 70.1 (O-CH2), 61.8 (O-CH2CH2N3), 50.7 (O-CH2CH2N3). 2-(2-(2-(2-Aminoethoxy)ethoxy)ethoxy)ethanol (13) The azide 12 (4.3 g, 19.6 mmol) was dissolved in methanol (50 mL) and treated with Pd/C (5m%, 200 mg). The suspension was flushed with argon prior to flushing with hydrogen. After stirring overnight at rt under hydrogen atmosphere the catalyst was filtered off and the filtrate was evaporated to give the title compound that was used without further purification for the next step. Formula: C8H19NO4. Yield: 3.8 g (19.6 mol, quant). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 3.55-3.75 (m, 12H, CH2), 3.52 (t, 2H, J = 4.9 Hz, CH2), 2.84 (t, 2H, J = 4.9 Hz, CH2). tert-Butyl 2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethylcarbamate (14) The amine 13 (3.4 g, 17.4 mmol) was dissolved in THF (30 mL) and treated with TEA (3.6 mL, 2.6 g, 26.0 mmol). A solution of boc2O (4.9 g, 22.6 mmol) in THF (10 mL) was added dropwise over 5 h to the reaction mixture that was subsequently stirred overnight at rt. The solvent was evaporated to give the crude product that was purified using flash chromatography (eluent: DCM/MeOH, 49:1) to give the title compound as a colorless viscous oil. Formula: C13H27NO6. Yield: 2.7 g (9.1 mmol, 52%). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 5.63 (s, 1H, NH), 3.68-3.78 (m, 4H, 2xCH2), 3.58-3.67 (m, 6H, 3xCH2), 3.53 (t, 2H, J = 5 Hz, CH2), 3.31 (m, 2H, CH2), 1.44 (s, 9H, tBu). ESI-MS: m·z-1 = 316.2 [M+Na]+(calc.: 316.2). 2-(2-(2-(2-(tert-Butoxycarbonyl)ethoxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (15) Alcohol 14 (4.1 g, 14 mmol), TsCl (3.4 g, 18 mmol) and TEA (3.9 mL, 2.8 g, 28 mmol) were mixed in dry DCM (25 mL) prior to cooling to 0 °C. Trimethylamine hydrochloride (0.7 g, 7 mmol) was added in one portion to the reaction mixture that was stirred at 0 °C for 3h and at rt overnight. The reaction mixture was concentrated to dryness and the residue purified using flash chromatography (eluent: DCM/MeOH, 95:5 +1% TEA). Formula: C20H33NO8S. Yield: 4.2 g (9.5 mmol, 68%). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 7.80 (d, 2H, J = 8.0 Hz, Ar-H), 7.34 (d, 2H, J = 8.0 Hz, Ar-H), 5.00 (s, 1H, NH), 4.16 (t, 7 2H, J = 4.9 Hz, CH2), 3.69 (t, 2H, J = 4.8 Hz, CH2), 3.57-3.63 (m, 8H, 4xCH2), 3.52 (t, 2H, J = 5.3 Hz, CH2), 3.30 (m, 2H, CH2), 2.45 (s, 3H, CH3), 1.43 (s, 9H, tBu). ESI-MS: m·z-1 = 470.2 [M+Na]+(calc.: 470.2). tert-Butyl 2-(2-(2-(2-(piperazin-1-yl)ethoxy)ethoxy)ethoxy)ethylcarbamate (16) Piperazin·6H2O (9.1 g, 47 mmol) and K2CO3 (1 g, 7 mmol) were dissolved in ACN (50 mL) and heated to 80 °C prior to adding tosylate 15 (2.1 g, 4.7 mmol) in ACN (10 mL) over 4 h to this suspension. The reaction mixture was stirred at 80 °C overnight, allowed to cool down to rt and the solvent evaporated. The residue was dissolved in DCM (50 mL) washed with sat. NaHCO3 solution (2x10 mL) dried over MgSO4 and the solvent evaporated. The crude product was purified using flash chromatography (eluent: DCM/MeOH, 95:5 +1% TEA) to give the title compound (1.25 g, 3.5 mmol, 74%). Formula: C17H35N3O5. Yield: 1.25 g (3.5 mmol, 74%). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 5.18 (s, 1H, NH), 3.57-3.70 (m, 10H, 5xCH2), 3.53 (t, 2H, J = 5.1 Hz, CH2), 3.31 (m, 2H, CH2), 2.92 (m, 4H, 2xCH2), 2.59 (t, 2H, J = 5.3 Hz, CH2), 2.51 (m, 4H, 2xCH2), 1.44 (s, 9H, tBu). ESI-MS: m·z-1 = 362.3 [M+H]+(calc.: 362.3). tert-Butyl 2-(2-(2-(2-(4-(4-bromophenyl)piperazin-1-yl)ethoxy)ethoxy)ethoxy)ethylcarbamate (17) Secondary amine 16 (3.9 g, 11 mmol), 1-bromo-4-iodo benzene (6.1 g, 22 mmol), K3PO4 (6.9 g, 33 mmol), CuI (3 g, 16 mmol) and ethylene glycol (911 µL, 1011 mg, 16 mmol) were dissolved in i PrOH (90 mL) and heated to 80 °C for 3 d. The solvent was evaporated and the residue dissolved in NaOH solution (100 mL). The aqueous phase was extracted with EE (5x20 mL) the combined organic layers washed with NaOH solution and dried over MgSO4. The solvent was evaporated to give the crude product that was purified using flash chromatography (eluent: EE+1% TEA) to give the title compound (4.4 g, 8.5 mmol, 78%). Formula: C23H38BrN3O5. Yield: 4.4 g (8.5 mmol, 78%). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 7.33 (d, 2H, J = 9.3 Hz, Ar-H), 6.78 (d, 2H, J = 9.1 Hz, Ar-H), 5.11 (s, 1H, NH), 3.57-3.70 (m, 12H, 6xCH2), 3.54 (t, 2H, J = 5.2 Hz, CH2), 3.31 (m, 2H, CH2), 3.18 (m, 4H, 2xCH2), 2.67 (m, 4H, 2xCH2), 1.44 (s, 9H, tBu). MALDI-MS: m·z-1 = 516.2 [M+H]+(calc.: 516.2). tert-Butyl 2-(2-(2-(2-(4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)piperazin-1-yl)ethoxy)ethoxy)ethoxy)ethylcarbamate (18) (Angew. Chem. 2010, 122, 7320-7322) Aryl bromide 17 (565 mg, 1.1 mmol), KOAc (215 mg, 2.2 mmol) and bis(pinacolato)diboron (362 mg, 1.4 mmol) were dissolved in ACN/H2O, 1:1 (1 mL). After 5 cycles of freeze-pump-thaw PdCl2(dppf) (80 mg, 0.11 mmol)was added prior to heating the reaction mixture for 60 min at 80 °C in the microwave (max. 20 W). After allowing to cool to rt EE (15 mL) was added and the organic phase washed with water (1x5 mL) sat. NaHCO3 solution (1x5 mL) and brine (1x5 mL). Evaporation of the solvent derived the crude product that was purified using flash chromatography (eluent: DCM/MeOH, 95:5 +1% TEA) to give the title compound (200 mg, 0.36 mmol, 32%). Formula: C29H50BN3O7. Yield: 200 mg (0.36 mmol, 32%). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 7.73 (d, 2H, J = 9.0 Hz, Ar-H), 6.87 (d, 2H, J = 8.5 Hz, Ar-H), 5.08 (s, 1H, NH), 3.91 (m, 2H, CH2), 3.56-3.85 (m, 12H, 6xCH2), 3.51 (m, 2H, CH2), 3.24-3.45 (m, 6H, 3xCH2), 3.10 (m, 2H, CH2), 1.44 (s, 9H, tBu), 1.32 (s, 12H, 4xCH3). 13C-NMR (150 MHz, CDCl3) : δ [ppm] = 156.1 (C=O), 151.4 (ArC), 136.3 (2xArC), 115.3 (2xArC), 83.5 (2xC3-C-O), 79.3 (Me3C), 70.2 (C-C-O), 70.1 (C-C-O), 65.6 (C-C-O), 60.4 (C-C-O), 56.6 (C-C-O), 52.3 (C-C-O), 56.6 (2xC-CN), 46.0 (2xC-C-N), 40.2 (C-C-O), 27.8 (3xCH3), 24.8 (4xCH3C-O). Exact mass (ESI): mz-1 = 564.38235 [M+H]+ (calc.: 564.38146). 8-Bromo-2(2-(4-(4-(2-(2-(2-(2-(tert-butoxycarbonyl)ethoxy)ethoxy)ethoxy)ethyl)piperazin-1yl)phenyl))quinoxaline (19) (WO 2008/148867 A2, PCT/EP2008057058) 2-Bromo-8chloroquinoxaline (9) (7.0 mg, 0.029 mmol), boronate 18 (16.0 mg, 0.029 mmol) and Na2CO3 (3.0 mg, 0.087 mmol) were dissolved in DMF (100 µL). This suspension was degassed with 5 cycles freeze8 pump-thaw prior to adding 5 mol% Pd(PPh3)4 (3.4 mg, 2.9 nmol). The reaction mixture was heated for 4 h at 100 °C in the microwave (max. 40 W). After allowing to cool to rt DMF was evaporated and the residue dissolved in DCM, filtered and the solvent evaporated to give the crude product that was purified using preparative RP-HPLC to give the desired product as its corresponding trifluoroacetate. Formula: C31H42BrN5O5. Yield: 5 mg (6.7 µmol, 24%). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 9.31 (s, 1H, Ar-H), 8.29 (d, 2H, J = 8.6 Hz, Ar-H), 8.07 (d, 1H, J = 8.4 Hz, Ar-H), 8.06 (d, 1H, J = 9.5 Hz, Ar-H), 7.56 (dd, 1H, J = 9.5, 8.4 Hz, Ar-H), 7.07 (d, 2H, J = 8.8 Hz, Ar-H), 5.07 (s, 1H, NH), 3.94 (m, 2H, CH2), 3.583.88 (m, 12H, 6xCH2), 3.53 (m, 2H, CH2), 3.24-3.45 (m, 6H, 3xCH2), 3.10 (m, 2H, CH2), 1.43 (s, 9H, tBu). 13 C-NMR (150 MHz, CDCl3) : δ [ppm] = 156.1 (C=O), 150.8 (ArC), 143.5 (ArC), 141.5 (ArC), 140.0 (ArC), 133.8 (ArC), 133.0 (ArC), 129.1 (2x ArC), 129.0 (ArC), 128.5 (ArC), 124.0 (ArC), 116.4 (2x ArC) 79.3 (Me3C), 70.3 (C-C-O), 69.9 (C-C-O), 69.8 (C-C-O), 69.6 (C-C-O), 65.7 (C-C-O), 65.1 (C-C-O), 56.6 (C-C-O), 51.8 (2xC-C-N), 45.3 (2xC-C-N), 40.3 (C-C-O), 27.8 (3xCH3). Exact mass (ESI): mz-1 = 644.24328 [M+H]+ (calc.: 644.24421). 8-(2,6-Difluorophenyl)morpholine)-2(2-(4-(4-(2-(2-(2-(2-(tert-butoxycarbonyl)ethoxy)ethoxy)ethoxy)ethyl)piperazin-1-yl)phenyl))quinoxaline (20) (WO 2008/148867 A2, PCT/EP2008057058) Bromoquinoxaline 19 (5 mg, 6.7 µmol), boronate 11 (2.7 mg, 8 µmol), SPhos (0.3 mg, 0.7 µmol) and K3PO4 (5.7 mg, 27 µmol) were dissolved in DMF (500 µL). After 5 cycles of freeze-pump-thaw Pd(OAc)2 (0.03 mg, 0.1 µmol) was added to the suspension prior to heating 2 h at 100 °C in the microwave (max. 40 W). After allowing the reaction mixture to cool to rt the solvent was evaporated. The residue was dissolved in DCM and the solids filtered off. After removal of DCM the crude product was purified using preparative RP-HPLC to give the title compound as its corresponding trifluoro acetate. Formula: C42H54F2N6O6. Yield: 1 mg (1.0 µmol, 15%). 1H-NMR (600 MHz, CDCl3): δ [ppm] = 9.37 (s, 1H, Ar-H), 8.18 (dd, 1H, J = 8.5, 1.7 Hz, Ar-H), 8.10 (d, 2H, J = 8.7 Hz, Ar-H), 7.82 (dd, 1H, J = 7.0, 1.6 Hz, Ar-H), 7.79 (dd, 1H, J = 8.5, 7.0 Hz), 7.54 (d, 2H, J = 9.3 Hz, Ar-H), 5.08 (s, 1H, NH), 4.42 (s, 2H, Benzyl-CH2), 3.99-4.06 (m, 4H, 2xCH2), 3.95 (m, 2H, CH2), 3.22-3.88 (m, 26H, 13xCH2), 1.42 (s, 9H, tBu). 13C-NMR (150 MHz, CDCl3) : δ [ppm] = 161.0 (2xArC), 156.1 (C=O), 151.0 (ArC), 150.6 (ArC), 143.0 (ArC), 142.8 (ArC), 139.2 (ArC), 139.1 (ArC), 136.5 (ArC), 130.7 (2x ArC), 130.6 (ArC), 130.3 (ArC), 128.7 (2xArC), 128.4 (ArC), 116.5 (2x ArC), 114.0 (2x ArC), 103.4 (ArC), 79.3 (Me3C), 70.3 (C-C-O), 69.9 (C-C-O), 69.8 (C-C-O), 69.6 (C-C-O), 65.7 (C-C-O), 65.1 (C-C-O), 63.8 (2xC-C-O), 56.6 (C-CO), 51.8 (2xC-C-N), 50.8 (BnC), 50.4 (2xC-C-N) 45.3 (2xC-C-N), 40.3 (C-C-O), 27.8 (3xCH3). Exact mass (ESI): mz-1 = 777.41365 [M+H]+ (calc.: 777.41457). 2-(2-(2-(2-(4-(4-(8-(3,5-Difluoro-4-(morpholinomethyl)phenyl)quinoxalin-2-yl)phenyl)piperazin-1yl)ethoxy)ethoxy)ethoxy)ethanamine (21) The carbamate 20 (1 mg, 1.0 µmol) was dissolved in 1 mL of a mixture of TFA, water and TIS (95:2.5:2.5) and stirred for 1 h at rt. The reaction mixture was coevaporated with isopropanol (2x10 mL) and dried in vacuum to give the title compound as its corresponding tritrifluoro acetate. Formula: C37H46F2N6O4, Yield: 0.92 mg (0.9 µmol, 90%) Analytical HPLC (Rt [min]): 2.25. Exact mass (ESI): mz-1 = 677.36183 [M+H]+ (calc.: 677.36214). 9 Synthesis of probe 3b: Supplementary Figure 3: Overview of the synthesis of probe 3b and its original inhibitor 4. 4-(4-(Benzyloxy)-3-methoxyphenyl)-2-chloropyrimidine (22) (Bioorganic & Medicinal Chemistry Letters 2009, 19, 5887–5892) 4-(Benzyloxy)-3-methoxyphenylboronic acid (300 mg, 1.162 mmol) and 2,4-dichloropyrimidine (260 mg, 1.744 mmol) were dissolved in 2M aqueous Na2CO3-solution (1.5 mL, 2.324 mmol). After addition of toluene (1.5 mL) the reaction mixture was degassed with 4 cycles of freeze-pump-thaw before Pd(PPh3)4 (130 mg, 0.12 mmol) was added to this suspension. The reaction mixture was heated to 100 °C for 120 min using mw irradiation (max 20 W). DCM (10 mL) and H2O (10 mL) were added prior to separation of the two layers. The organic phase was washed with sat. NaHCO3-solution (3x3 mL), brine (3x3 mL), dried over MgSO4 and the solvent evaporated. Residual dichloropyrimidine was removed via short way distillation. The crude product was used as obtained in the next step. Formula: C18H15ClN2O2. Yield: 379.7 mg (1.162 mol, quant.). LC-MS (ESI): Rt = 9.04 min, mz-1 = 327.3 (calc.: 327.1). tert-Butyl piperazine-1-carboxylate (23) Piperazine (1 g, 11.6 mmol) and amberlyst-15 (15 %, 150 mg) were dissolved in DCM (50 mL) and cooled to 4 °C. A solution of Boc2O (2.3 g, 10.5 mmol) in DCM (25 mL) was added dropwise over 2 h to this suspension. After allowing to warm up to rt the reaction mixture was filtrated and evaporated. The crude product was purified using flash chromatography (eluent: DCM/MeOH 95:5 +1 % TEA) to give the title compound. Formula: C15H23N3O2. Yield: 1.17 g (6.26 mmol, 54 %). 1H-NMR (500 MHz, CDCl3): δ [ppm] = 3.34-3.46 (m, 6H, 6xH-CH), 2.83 (m, 2H, 2xH-CH), 1.46 (s, 9H, tBu). tert-Butyl 4-(4-aminophenyl)piperazine-1-carboxylate (24) Secondary amine 23 (1.17 g, 6.26 mmol), 4-iodobenzeneamine (920 mg, 4.2 mmol), K3PO4 (1.78 g, 8.2 mmol), CuI (400 mg, 2.1 mmol) and ethylene glycol (463 µL, 509 mg, 8.2 mmol) were dissolved in iPrOH (90 mL) and heated to 80 °C for 3 d in a sealed reaction tube. The solvent was evaporated and the residue purified using flash 10 chromatography (eluent: DCM/EE 1:1) to give the title compound. Formula: C15H23N3O2 Yield: 859 mg, 3.1 mmol, 74%, LC-MS (ESI): Rt = 4.32 min, mz-1 = 278.3 (calc.: 278.2). 2-(2-(2-(2-Azidoethoxy)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (25) Alcohol 8 (900 mg, 4.1 mmol), TsCl (1010 mg, 5.3 mmol) and TEA (1137 µL, 830 mg, 8.2 mmol) were dissolved in dry DCM (20 mL) prior to cooling to 0 °C. Trimethylamine hydrochloride (118 mg, 1.23 mmol) was added in one portion to the reaction mixture that was stirred at 0 °C for 3h and at rt overnight. The reaction mixture was concentrated to dryness and the residue purified using flash chromatography (eluent: DCM/EE 1:1). Formula: C15H23N3O6S Yield: 1082 mg (2.9 mmol, 71%). LC-MS (ESI): Rt = 1.09 min, mz-1 = 373.5 (calc.: .374.1). tert-Butyl 4-(4-(4-(4-(benzyloxy)-3-methoxyphenyl)pyrimidin-2-ylamino)phenyl)piperazine-1-carboxylate (26) (Bioorganic & Medicinal Chemistry Letters 2009, 19, 5887–5892) Aryl chloride 22 (40 mg, 0.123 mmol) and amine 24 (34 mg, 0.123 mmol) were dissolved in 1,4-dioxane (1 mL) and treated with pTsOH monohydrate (7 mg, 0.04 mmol). The resulting suspension was heated to 80 °C for 10 h and the solvent evaporated to give the crude product that was purified via preparative RP-HPLC to give the title compound. Formula: C33H37N5O4. Yield: 24 mg (0.042 mmol, 34 %). LC-MS (ESI): Rt = 9.60 min, mz-1 = 568.4 (calc.: 568.3). 2-Methoxy-4-(2-(4-(piperazin-1-yl)phenylamino)pyrimidin-4-yl)phenol (27) The protected aryl compound 26 (24 mg, 0.042 mmol) was dissolved in a solution of TFA/H2O/TIS 95/2.5/2.5 (1.5 mL) and stirred at rt for 120 min. Volatile substances were evaporated and the residue purified via preparative RP-HPLC to give the desired product as its corresponding trifluoroacetate. Formula: C21H23N5O2. Yield: 9.9 mg (0.0201 mmol, 48 %). LC-MS (ESI): Rt = 3.81 min, mz-1 = 378.5 (calc.: 378.2). 4-(2-(4-(4-(2-(2-(2-(2-Azidoethoxy)ethoxy)ethoxy)ethyl)piperazin-1-yl)phenylamino)pyrimidin-4-yl)2-methoxyphenol (28) The secondary amine 27 (3 mg, 0.0061 mmol) and TEA (200 µL, 240 mg, 1.61 mmol) were dissolved in dry DCM (1 mL) and treated with tosylate 25 (301 mg, 0.805 mmol). The resulting reaction mixture was heated to reflux for 6 h. After evaporating the solvent the crude product was purified via RP-HPLC to give the title compound as its corresponding trifluoroacetate. Formula: C29H38N8O5. Yield: 4 mg (0.006 mmol, 95 %). 1H-NMR (600 MHz, MeOD): δ [ppm] = 8.24 (d, 1H, J = 5.91 Hz, Ar-H), 7.84 (d, 1H, J = 1.8 Hz, Ar-H), 7.72 (dd, 1H, J = 2.1, 8.3 Hz, Ar-H), 7.60 (d, 2H, J = 9.0 Hz, Ar-H), 7.37 (d, 1H, J = 6.3 Hz, Ar-H), 7.11 (d, 2H, J = 9.0 Hz, Ar-H), 6.93 (d, 2H, J = 8.4 Hz, ArH), 3.95 (s, 3H, CH3), 3.90 (m, 2H, CH2), 3.70-375 (m, 4H,2xCH2), 3.65-3.69 (m, 8H, 4xCH2), 3.37-3.50 (m, 8H, 4xCH2), 1.31 (m, 4H, 2xCH2). LC-MS (ESI): Rt = 4.63 min, mz-1 = 579.4 (calc.: 579.3). 4-(2-(4-(4-(2-(2-(2-(2-Aminoethoxy)ethoxy)ethoxy)ethyl)piperazin-1-yl)phenylamino)pyrimidin-4yl)-2-methoxyphenol (29) The azide 28 (2 mg, 0.0035 mmol) was dissolved in MeOH (1 mL) and treated with TEA (200 µL, 240 mg, 1.61 mmol) and 1,3-propanedithiol (200 µL, 216 mg, 2.0 mmol). The reaction mixture was stirred overnight and the solvent evaporated prior to preparative RP-HPLC purification. The title compound was obtained as its corresponding ditrifluoroacetate. Formula: C29H40N6O5. Yield: 2.7 mg (0.0035 mmol, quant). 1H-NMR (600 MHz, MeOD): δ [ppm] = 8.26 (d, 1H, J = 5.9 Hz, Ar-H), 7.83 (d, 1H, J = 2.0 Hz, Ar-H), 7.71 (dd, 1H, J = 8.4, 2.0 Hz, Ar-H), 7.61 (d, 2H, J = 9.0 Hz, Ar-H), 7.35 (d, 1H, J = 6.0 Hz, Ar-H), 7.09 (d, 2H, J = 9.0 Hz, Ar-H), 6.92 (d, 1H, J = 8.3 Hz, ArH), 3.95 (s, 3H, CH3), 3.90 (m, 2H, CH2), 3.65-3.80 (m, 18H, 9xCH2), 3.47 (m, 2H, CH2), 3.12 (m, 2H, CH2). 13C-NMR (125 MHz, CDCl3): δ [ppm] = 167.1 (ArC), 157.5 (ArC), 152.1 (ArC), 151.0 (ArC), 147.8 (ArC), 146.1 (ArC), 127.5 (ArC), 122.5 (2xArC), 122.0 (ArC), 121.7 (ArC), 117.3 (2xArC), 115.0 (ArC), 11 110.6 (ArC), 106.2 (ArC), 70.0 (CH2), 69.9 (7xCH2), 66.2 (CH2), 63.8 (CH2), 55.9 (CH2), 55.0 (CH3), 39.2 (CH2). LC-MS (ESI): Rt = 3.42 min, mz-1 = 553.4 (calc.: 553.3). Exact mass (ESI): mz-1 = 553.31317 (calc.: 553.31329). 2-Methoxy-4-(6-(4-morpholinophenylamino)pyrazin-2-yl)phenol (30) (Bioorganic & Medicinal Chemistry Letters 2009, 19, 5887–5892) Aryl chloride 22 (106 mg, 0.324 mmol), 4morpholinobenzenamine (58 mg, 0.324 mmol) and pTsOH∙H2O (19 mg, 0.097 mmol) were dissolved in 1,4-dioxane (3 mL) and heated to reflux for 4 h. Volatile substances were subsequently evaporated to give the crude substitution product that was treated with 95 % aqueous TFA (10 mL) and stirred for 8 h at 60 °C. After the reaction mixture reached ambient temperature the solvent was evaporated and the crude product purified using preparative HPLC to give the title compound. Formula: C21H22N4O3, Yield: 78 mg (0.206 mmol, 64 %, over two steps), 1H-NMR (500 MHz, CDCl3): δ [ppm] = 11.79 (s, 1H, NH), 8.09 (d, 1H, J = 6.3 Hz, ArH), 7.74 (d, 1H, J = 2.1 Hz, ArH), 7.63-7.68 (m, 3H, 3xArH), 7.17 (d, 1H, J = 6.7 Hz, ArH), 7.04 (d, 1H, J = 8.3 Hz, ArH), 6.98 (d, 2H, J = 8.5 Hz, 2xArH), 3.98 (s, 3H, CH3), 3.90 (m, 4H, 2xCH2), 3.18 (m, 4H, 2xCH2). 13C-NMR (125 MHz, CDCl3): δ [ppm] = 164.3 (ArC), 164.0 (ArC), 153.5 (ArC), 151.2 (ArC), 148.3 (ArC), 147.1 (ArC), 129.9 (ArC), 126.5 (ArC), 123.2 (ArC), 123.0 (2xArC), 116.2 (2xArC), 115.1 (ArC), 110.3 (ArC), 105.1 (ArC), 66.7 (2xCH2), 56.0 (CH3), 49.8 (2xCH2). MS (MALDI): mz-1 = 378.8 (calc.: 379.2). 12 Binding profile of probe 3 Supplementary Table 3: Kinases identified in all replicates in the inhibitor affinity purification experiment using probe 3b and MV4-11 cell lysate. JAK isoforms JAK1, JAK2 and Tyk2 are highlighted in red. 13