Supplementary Information (docx 22K)
advertisement
")
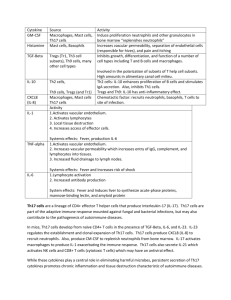
Supplemental Experimental Procedures Flow cytometry analysis of ICAM-1 expression on Treg and Tconv cells, CD11b and IL-10 receptors on neutrophils. ICAM-1 expression was analyzed on unstimulated, LPS- or CD3/CD28-stimulated Treg or Tconv cells using flow cytometry. T cells were incubated for 0-48 h and then stained with mAbs. CD11b expression was analyzed on unstimulated neutrophils or neutrophils incubated with LPS- or CD3/CD28-stimulated Treg cell. For immunostaining, ICAM1-PE (LB-2, BD Pharmingen, San Jose, CA, US), CD11b-PE (D12, BD Pharmingen) and respective mouse isotype control was employed (IgG2a-PE, DAK-G05, Daco A/S, Glostrup, Denmark). IL-10 receptors expression was analyzed on unstimulated, LPS (100 ng/mL) or hIL-10 (5 ng/mL) stimulated neutrophils (5 h). For immunostaining, mouse anti-IL10RA mAb (2 g/mL, 37607.11, Abcam, Cambridge, UK) and secondary antibody conjugated with PE (1:10, goat anti-mouse, Daco A/S) were used. As a negative secondary antibody control, PE-conjugated mouse IgG2a was used (DAK-G05, Daco A/S). After 30 minutes of incubation, samples were washed, fixed with 1% PFA and analyzed (LSRII, BD, San Jose, CA, US). Microscopy. For quantitative immunofluorescence analysis, cells cultured on Biocoat 96-well black/clear plate coated by poly-D-Lysine cellware (Becton Dickinson Labware, San Jose, CA, US ) or cells transferred from Ubottom plates to gelatin-coated microscope slides by cytospine were perfused with PBS containing 4% (w/v) paraformaldehyde for 20 min at 21C. Fixed cells were extensively washed with PBS and blocked with 10% rabbit normal blocking serum (Santa Cruz Biotechnology, Santa Cruz, CA, US) supplemented with 3% TritonTM X-100 (Sigma-Aldrich, St. Louis, MO, US) for 45 minutes at 21C. Next, cells were washed and incubated with unlabeled 5 g/mL anti-human IL-10 mAbs (rat, clone JES3-9D7, Invitrogen, Waltham, MA, US) or rat IgG2b-negative isotype control (Invitrogen) in PBS supplemented with 1.5% normal blocking rabbit serum and 0.3% Triton X-100 and 0.01% sodium azide, overnight at 4C in a wet chamber. Subsequently, cells were washed in PBS three times and secondary fluorescent antibody (rabbit pre-adsorbed polyclonal TRITC, Abcam, 1:100) supplemented with 1.5% normal blocking rabbit serum and 0.3% Triton X-100 and 0.01% sodium azide were added for 1 h at 21C. For double staining, cells were in parallel incubated with primary direct antibody CD15-FITC (MMA, 0.00625mg/mL, BD Pharmingen) or CD18-FITC (L130, 0.00625 mg/mL, BD Pharmingen) and washed three times in PBS. Alternatively, neutrophils were incubated in parallel with unlabeled 5 g/mL anti-human IL-10 mAbs (rat, JES39D7, Invitrogen) and 1 g/mL Annexin V (H3, Santa Cruz Biotechnology) or rat IgG2b negative isotype control for IL-10 (Invitrogen). Rabbit pre-adsorbed polyclonal TRIC (1:100; Abcam) and goat pre-adsorbed polyclonal FITC (1:100; Abcam) secondary fluorescent antibodies were used. For fluorescent DNA nuclei staining, Hoechst 33342 (fixed cells on the 96-well plate; 1 g/mL in PBS, Sigma Aldrich) or DAPI (fixed cells on the micro slides; 1.5 g/mL UltraCruz Mounteining Medium, Santa Cruz Biotechnology) were used. Images were acquired using confocal microscope BD PathwayTM Bioimager 850 with AttoVision 1.5.3 software (BD Bioscience, San Jose, CA, US). For quantitative analysis of IL-10-positive neutrophils, sixteen sites per well (Biocoat 96-well black/clear plate) were analyzed using Olympus 20×0.75 NA objective. To measure fluorescence intensity, cells were cytospined (300xg, 10min) on gelatin-coated microscope slides, and sixteen sites per microscope slide were analyzed using Olympus PlanApo N 60x1.42 oil objective and IPLab Pathway 4.0 software (BD Bioscience, San Jose, CA, US). Fluorescence intensity was determined as the Average fluorescence (avg. area), the sum of the fluorescence from all segments divided by the number of segments.1 The average fluorescence was calculated using at least one hundred single cells taken from four independent experiments. The level of baseline fluorescence was established individually for each experiment. Nonspecific fluorescence (signal noise) was electronically diminished to the level when nonspecific signal was undetectable. The intracellular localization of IL-10 was analyzed by confocal microscopy (Nikon D-Eclipse C1) using EZ-C1 v. 3.6 software. Image analysis and volume rendering was performed using NIS-elements Ar software (Nikon, Tokyo, Japan). The visualization of live cell interactions between neutrophils and LPS-activated Treg cells was performed on 8-well glass chamber slides (Nalge Nunc International, Waltham, MA, US). During the course of 21 hours, neutrophil-Treg cell interactions were imaged at five time points (1, 3, 5 and 7 hour intervals) with a Zeiss Axiovert 200 inverse microscope with a Zeiss Plan-apochromat 63x/1.43 oil differential interference contrast objective (Göttingen. Germany). Additionally, after 21-h incubation cells were fixed and labeled with CD18-FITC, DNADAPI, IL-10-TRITC antibodies as described above, and images were acquired using Zeiss Axiovert 200 inverse microscope with different settings of filters. Western blot analysis. After incubation of neutrophils with unstimulated or stimulated Treg cells at a ratio of 50:1 for 21 h, the cells were centrifuged (1min, 12000xg) and lysed in the ice-cold RIPA lysis buffer (1%Triton X-100, 20mM Tris, 150mM NaCL, pH 7.4) containing a 1mM PMSF, 5 mM NaF, 2 mM activated sodium ortovanadate, 1mM EGTA, 1mM EDTA, 1% protease inhibitor cocktail for 30 min on ice (all reagents were purchased from Sigma-Aldrich). The protein concentration in the lysates was determined using an RC DC Protein Assay Kit (BioRad, Hercules, CA, US). Cell lysates containing the same amount of proteins were run on a 10% SDS-PAGE minigel and the proteins were transferred to PVDF membranes (BioRad) at 35 mA, 4C for 18 hours. The membranes were blocked with 1% BSA and 1% PEG 3500 with 10 mM NaF in 2 x TBS-Tween 20 for 2 hours (all reagents from Sigma-Aldrich) and then incubated at room temperature for 2 hours with primary mouse anticytochrome c (7H8.2C12, BD Pharmingen) (1:500) and mouse IgG anti-β actin (Sigma-Aldrich) (1:4000) antibodies. The membranes were then washed 5 times in TBS-Tween 20, incubated with HRP-conjugated goat anti-mouse IgG (1:1000, Sigma-Aldrich) at room temperature for 1 hour and washed. The proteins were visualized using an immunoblotting-enhanced chemiluminescence system (ECL, Santa Cruz Biotechnology). The densitometric scanning of the blots and the analysis of the visualized bands were performed using a Fluoro-Chem Multi Image FC Cabinet (Alpha Innotech Corporation, San Leandro, CA, US) and analyzed with AlphaEaseFC software, version 3.1.2. ELISA. IL-10, was measured in supernatants of 5-h incubations of neutrophils with unstimulated or stimulated Treg cells using ELISA Kits (Bender MedSystems, Vienna, Austria). The detection limit was 0.39 pg/mL. RNA isolation and real-time quantitative PCR. Unstimulated or stimulated Treg cells were cocultured with neutrophils at a ratio of 1:50 for 5 h. T cells were then removed with CD2 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). Total RNA was isolated with an RNeasy Mini Kit (Qiagen, Limburg, Netherlands) according to the manufacturer’s instructions, and treated with RNase-free DNase I (Qiagen) to remove contaminating genomic DNA. Following this, 1 µg of total RNA was reverse transcribed to cDNA with random hexamers and M-MMLV reverse transcriptase using a RT2 First Strand Kit (Qiagen). Real time RT-PCR was performed to assess the expression levels of mRNA for IL-10, HCAR3 (Puma), NCF-2 (NOXA), GAPDH and -actin using gene specific primer sets (IL-10; Refseq Accession number NM_000572, HCAR3; NM_006018, NCF-2; NM_000433, GAPDH; NM_002046, and -actin; ACTB, NM_001101). Reaction was performed using RT 2 SYBR Green ROX qPCR Mastermix to which set of gene specific primers was added. cDNA synthesized from Human XpressRefTM Universal Reference Total RNA (SA Bioscience, Limburg, Netherlands) was used as a positive control. Real time RT-PCR was performed using 7500 Real Time PCR Applied Biosystem (Waltham, MA, US), and the cycling conditions were as follows: 95C, 15 min; 40 cycles of three stages: 95C for 30 sec, 55C for 30 sec and 72C for 30 sec. The quality of the reaction was verified by melting curve analysis, and the relative expression of the genes was determined with software supplied by Applied Biosystem. The average expression levels of housekeeping genes, the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and -actin (ACTB), were used for the normalization of the cDNA samples. Multiple Gene Profiling Microarray. Gene expression was analyzed using Human apoptotic RT2 Profiler PCR Array (SuperArray, USA). RT-PCR was performed according to the manufacturers recommendations and tested cDNA was amplified in the presence of 84 specific primers (RefSeq accession numbers presented in the supplementary Table 1), coated in 96-well microtiter plates. Reaction was performed on an Applied Biosystem 7500 Real Time PCR. Data from real-time PCR were calculated using the Ct method and the PCR Array Data Analysis Template v3.0 (SuperArray, Limburg, Netherlands) as previously described.2 Chromatin immunoprecipitation (ChIP) assays. Chromatin immunoprecipitation was performed using EZ-Magna ChIP™ A/G Kit (Millipore, Darmstadt, Germany) according to the manufacturer’s protocol. To better visualize changes in chromatin reorganization, neutrophils were incubated with Treg cells for 21 h, and then negatively separated by magnetic beads. Briefly, neutrophils were fixed with formaldehyde (1% PBS, 10 min., room temp.) to generate protein-DNA crosslinks. Neutrophils (4x106 cells) were then lysed and chromatin was released from the nuclei and sheared by sonication at 85% amplitude by 0.5 sec. on - 1 sec. off pulses (Ultrasonic Homogenizer Sonoplus mini 20, Banderin, German) to obtain DNA fragments of 200-1000 bp. The efficiency of sonication and the DNA analysis was performed using commercially available Agilent High Sensitivity DNA kit (2100 Bioanalyzer, Agilent 2100 expert software, Santa Clara, CA, US). 3 L of anti-trimethyl-Histone H3 antibodies (H3K4me3, clone MC315, ChIP qualified antibody, Millipore), 2 L of anti-acetyl-Histone H3 antibodies (Lys4) (H3Ac Lys4, ChIP qualified antibody, Millipore), 1 g of mouse anti-CTD region of RNA Polymerase II antibodies (positive control, Millipore) and 1 g of normal mouse IgG antibodies (negative control for non-specific immunoselection of chromatin by immunoglobulins, Millipore) were conjugated to Protein A/G magnetic beads and used to immunoprecipitate and isolate the crosslinked proteins with the magnetic separators (Magna GrIP Rack, Millipore). The formaldehyde crosslinking was reversed by heat treatment, and the DNA associated with each protein was purified and used to perform RT-PCR (SybrGreen, Applied Biosystems) for the human IL-10 promotor genomic locus specific primer forward 5’GAGTGTCCCTGCTGGTCTGTAG3’, reverse 5’CAAGCCTTGTCTGAGATGATCC3’ (Invitrogen). To establish the background levels of ChIP experiments, the histone modifications were quantified also at the promoter of prolactin (silent in myeloid cells) using specific primers forward 5’AGGGAAACGAATGCCTGATT3’, reverse 5’GCAGGAAACACACTTCACCA3’).3 REFERENCES 1. 2. 3. Dima, A.A. et al. Comparison of segmentation algorithms for fluorescence microscopy images of cells. Cytometry A. 79, 545-559 (2011). Hansel, N.N. et al. Analysis of CD4 T-cell gene expression in allergic subjects using two different microarray platforms. Allergy 63, 366–369 (2008). Tamassia, N. et al. Cutting edge: An inactive chromatin configuration at the IL-10 locus in human neutrophils. J. Immunol. 190, 1921-1925 (2013).