Drugs used for the immobilization, capture, and translocation of wild
advertisement

Drugs used for the immobilization, capture, and translocation of wild animals
G.E. Swan
1 General considerations
1.1 Introduction
Decisions made concerning the application of drugs for the immobilization, capture and translocation of wild animals
depend on a thorough knowledge of the nature of the drugs being used. Criteria applicable to the choice of a drug for a
particular application include:
•Availability.
•Safety - for animal and user.
•Previous results in the particular species under consideration.
•The duration of effect required.
•The need for an antidote.
•The legislative implications of using the drug.
In order to shed light on these and other criteria, this chapter reviews the drugs most readily obtained and most commonly
used in the field of wildlife management in southern Africa.
Anaesthetic agents, opioids, hypnotics, sedatives, tranquillizers (neuroleptics) and neuromuscular blocking drugs are the
groups of drugs most commonly used for the immobilization and capture of wild animals, and during subsequent
translocation of these animals. Other drugs are used for chemical reversal of central nervous system and neuromuscular
depressant effects, and for cardiovascular and respiratory support following or during chemical restraint. Immobilizing
drugs are all potentially dangerous to humans and animals and should, therefore, be handled and used with the utmost
caution. Strict national legal control measures must be complied with.
Immobilization, or the chemical restraint, of wild animals has replaced, or is used in conjunction with, various types of
physical restraint and capture methods. The availability of suitable drugs has dramatically improved the safety and
cost-effectiveness of the handling, capture, transportation, and adaptation following translocation, of wild animals.
Before the advent of suitable drugs, the capture and transportation of many wild animals, particularly the larger species
such as elephant and rhino, was extremely difficult and hazardous. With certain antelope species, physical restraint was
formerly virtually impossible due to the phenomenon of stress-induced mortality.
Growing public awareness of animal welfare issues will in future lead to increasingly stringent ethical controls on the
handling and treatment of non-domesticated animals. In particular it will lead to, amongst other things, the increased use
of chemical immobilization and stress management.
1.2 History
The capture of bears with an oral mixture of honey and strong spirits by Winckell in 18201 is probably the first
documented case of the use of chemicals to restrain wild animals. From then on, a number of oral mixtures were used
with variable success2,3,4,5,6,7,8,9. Oral administration of anaesthetics and tranquillizers for the capture or handling of
wild animals has been described as virtually impossible10 and frustrating11. The length of the induction period and the
degree of sedation or immobilization achieved when using this route of administration are highly unpredictable8,12.
The true birth of chemical restraint of wild animals coincides with the use of the first 'drug dart' by Hall and co-workers in
195313. Neuromuscular blocking agents, ranging from early synthetic curare14,15, nicotine salicylate15,16,17,4,11,18
and strychnine salts1 were the first agents successfully used to provide chemical restraint. However, a major drawback of
these early drugs was that their safety margin, particularly when used in larger animals, was too narrow. A significant
breakthrough was made in 1960 by Harthoorn19. Working with the ranger team of the Natal Game and Fish Preservation
Board he pioneered the use of a mixture of morphine or synthetic morphine, hyoscine, and various tranquillizers20. The
development and use of newer, extremely potent synthetic and semi-synthetic opioids (fentanyl, etorphine, carfentanil),
cyclohexylamines, phenothiazine derivatives, and alpha-2-agonists, then followed. These groups of agents, alone or in
different combinations, now form the basis of non-domestic animal immobilization1,9,21,22,23,24,25.
The most recent major advance in the translocation of wildlife has been the adoption of human long-acting tranquillizers
for use in this field. These drugs have significantly reduced the numbers of animals lost during long-distance
transportation, while in captivity, and following introduction into new habitats (see The Use of Long-acting
Tranquillizers in Captive Wild Animals, this Section).
1.3 Legal control
The information given here on the legal control of drugs used in the wildlife field pertains specifically to South Africa.
Similar legislation applies in other countries, and the reader is urged to familiarize him- or herself with the legal
implications of using drugs in the country in which he or she is active.
All drugs used for the immobilization, capture, and translocation of wild animals in South Africa are registered or
controlled under the Medicines and Related Substances Control Act No. 101 of 1965. The Act provides that all
medicinal substances be placed into nine schedules, representing four main groups, according to safety profile and use.
Scheduling determines the availability and type of control placed on a particular medicinal substance.
In terms of the Act:
•Unscheduled (over the counter (OTC)) medicines may be
supplied through any retail outlet.
•Schedule 1 and 2 medicines are available through a pharmacist.
•Schedule 3 to 7 medicines are prescription medicines.
•Schedule 8 and 9 medicines are prohibited substances that
require a special permit for acquisition and use.
In general, the prescription restrictions and requirements, and the record-keeping procedures for holding and handling
medicines, become more stringent the higher the schedule becomes. The relevant control measures will be discussed
separately under each of the groups of drugs used in wild animals. Some general principles include the following.
•Prescription medicines (i.e., Schedules 3 to 7) used in animals are available only through a veterinarian, or
through a pharmacist on a veterinary prescription.
•A joint Medicines Control Council (MCC) of Act 101 of 1965 and S.A. Veterinary Council policy outlines the
conditions under which a veterinarian may prescribe a narcotic or psychotropic medicine to a non-veterinarian
for use in wild animals26. In terms of this policy: a prescription by a veterinarian for the use of these products
may only be issued to a bona fide client; the client must be fully informed of the correct use and potential
dangers of the products; and, an affidavit must be signed by the client to this effect.
•In terms of an ethical rule of the Veterinary Council, a veterinarian may not prescribe medicines for veterinary
purposes or procedures for which he/she is not experienced or qualified.
•Medical practitioners and dentists (who are entitled to prescribe scheduled medicines) are, in the same way as
veterinarians, prohibited in terms of Section 22a of Act 101 of 1965 to prescribe, sell, or use these drugs for
any purpose other than as required in their normal line of professional activity.
•For persons other than those legally entitled to do so, it is an offence to have a Schedule 5 or higher medicine
in their possession without a prescription.
•An application for a permit to obtain and use Schedule 8 and 9 substances is made to the Registrar of
Medicines, Act 101 of 1965. On recommendation by the MCC, the permit may be issued by the Director
General of the Department of National Health and Population Development. Schedule 8 drugs may only be
approved for research or analytical purposes.
•Dangerous, dependence-producing substances (such as the opioids and phencyclidine) used for restraint of
wild animals are, in addition to national legislation, controlled in terms of the international 'Single Convention
on Narcotic Drugs of 1961' and the 'Convention on Psychotropic Substances of 1971'. South Africa is a
party to both of these conventions. These conventions control the importation and availability of dangerous
dependence-producing substances and psychotropic substances at an international level. All parties to these
Conventions have undertaken to apply the measures of control laid down in each Convention. The Department
of National Health and Population Development in South Africa is responsible to the International Narcotics
Control Board in this respect.
Residues in food originating from animals are controlled in terms of the Act on Foodstuffs, Cosmetics and
Disinfectants (Act 54 of 1972) and the Act on Hygiene at Animal Slaughter, Meat and Animal Products (Act 87 of
1965). These Acts determine that there should be no drug residues in the meat of animals at the time of slaughter. In most
cases, no withdrawal periods for drugs used in wild animals have been determined. It is therefore incumbent upon the
veterinarian involved to prescribe an adequate withdrawal period for any drug used in wild animals that are intended for
slaughter. A period of at least three months has been suggested for products of which the withdrawal period has not been
determined. Alternatively, if the biological half-life of a product is known for a particular species, then a withdrawal
period of 5-6 half-lives for that species could be used.
The importation and use of drugs not registered in South Africa requires approval under Section 21 of Act 101 of 1965.
An application for use of such a drug in a patient or for trial purposes must be made to the Registrar of Medicines and
must be approved by the MCC.
Most veterinary drugs used for wildlife are not registered for this particular purpose in South Africa. In addition,
many of these drugs are not even registered for veterinary use, but are only intended for human health. A veterinarian may
prescribe such drugs for extra-label use in wild animals, but takes full legal responsibility therefor.
In 1991 the Medicines and Related Substances Control Amendment Act No. 94 of 1991 (Act 94/91) was passed by
Parliament. One of the objectives of this act was to make provision for the control of medicines through regulations rather
than through the rigid control of Act 101 of 1965. It has now become possible to supply Schedule 8 and 9 substances for
reasons other than for research and analytical purposes. New regulations to the Act are currently being prepared, with
enactment envisaged in 1993. Changes in the schedule structure, and consequently a change in the availability of
medicines, is anticipated. The new act also provides for a person, organization, or group of persons to possess and use
immobilizing drugs in Schedules 5 to 8. This is subject to conditions prescribed in regulations and specified in a Notice to
the Act. These conditions will be included in the regulations projected for 1993.
1.4 Human safety
Wildlife management personnel, technical staff, and veterinarians, all with widely varying degrees of expertise in
handling toxic substances, are involved in the handling and use of immobilizing drugs at doses that exceed levels
potentially fatal to humans27. Neuromuscular blocking agents and opioids are particularly dangerous, leading to acute
respiratory failure. At least one fatality and numerous exposures have been reported28,29,30,31,32,33,34. Many more
accidental exposures may occur annually, but are not reported27.
Drugs can enter the circulation by absorption through the intact skin and mucous membranes, sub-cutaneously through
cuts, scratches or injections, and intramuscularly or intravenously by direct injection. In most cases, incidents can be
ascribed to negligence during the loading of darts and syringes or during the preparation of solutions, or to the careless
handling of syringes, darts, or power injectors27. The loading of darts during helicopter capture procedures is particularly
hazardous.
To prevent accidental exposure, the following general precautions should be observed during the handling and
use of any immobilizing agent.
•Ensure the availability of a suitable first-aid kit equipped with resuscitation equipment and appropriate
quantities of suitable antidotes. When working with drugs such as etorphine or other extremely potent opioids,
it may be advisable to prepare the antagonist before handling the drug.
•Never work alone when mixing drugs and loading syringes or darts. All persons involved must be adequately
trained and experienced to administer first-aid and the required antidote when necessary.
•Wear rubber gloves and glasses when mixing powdered drugs into solution, or when handling large quantities
of solutions.
•Avoid spraying, squirting, or spilling drugs when loading. The use of luer-lock syringes and an extra needle to
relieve pressure when extracting a drug from a vial is useful in this respect.
•In the case of contact with skin or mucous membranes, wash immediately using copious quantities of water.
Should injection into a limb occur, the use of a pressure bandage will limit the quantity of drug absorbed and
the toxic reactions that could result.
•Treat all dart guns and projectile syringes with the utmost care, as in the case of a loaded firearm.
The effects of poisoning in humans, and the antidotes available for the various immobilizing drugs, are discussed under
each group of drugs. Many of the drugs used in animal immobilization are unknown to most physicians. It is, therefore,
advisable to inform your local medical community of the type of drugs that you are using, and of the indicated treatments
and antidotes.
Respiratory failure leading to hypoxia, hypotension, and cardiac arrest are the cause of death in all forms of acute drug
poisoning. Whilst antagonists may delay the onset of symptoms in some cases, there are no antidotes for depolarizing
peripheral muscle relaxants, and in such cases artificial ventilation is essential. Prompt use of cardiopulmonary
resuscitation (CPR) will vastly improve the chances of survival of anyone having cardiopulmonary arrest. Every second
that the brain is deprived of oxygen leads to further brain damage, which may become irreversible if ventilation is delayed
for too long.
If sufficiently well trained and experienced personnel are available, the insertion of an endotracheal tube will protect the
airway against vomitus and allow ventilation to be carried out more easily. Persons inexperienced in intubation are best
advised to continue with mouth-to-mouth resuscitation or to use an Ambu® resuscitation bag and mask.
All persons that have been accidentally injected with a dangerous drug should be transferred to a hospital as soon as
possible. During transportation it is essential that the patient remain under close observation so that further resuscitation
measures can be applied if necessary. A full description of all drug treatments and CPR procedures administered must be
supplied to the attending physician(s) before or on arrival at the hospital.
For details on how to deal with accidental administration of immobilizing drugs, see Prevention and Management of
Capture Drug Accidents, this Section.
1.5 Characteristics of an 'ideal' immobilization drug
The characteristics of an ideal immobilization drug for darting wild animals can be summarized as follows1,6,25.
•An effective dose should not exceed the quantity that can be carried in an appropriately-sized, preferably less
than 3 ml, dart.
•Suitable stability.
•Rapid absorption into the systemic circulation.
•Rapid onset of action resulting in sufficient immobilization.
•Duration of effects should be long enough.
•A wide margin of safety (at least 3-fold).
•Availability of a reliable antidote.
•Rapid elimination from the body.
•No drastic effect on pregnant animals.
•No permanent damage to the animal.
•Should not cause tissue irritation.
•Minimum effects on cardiorespiratory function.
•Recovery and induction periods should be calm.
•Minimum risk to personnel handling the drug.
Harthoorn29 also suggested that the ideal immobilizing drug should reduce the animal's awareness of its surroundings to
minimize fear, distress, and pain.
1.6 Classification
Drugs used for the immobilization, capture, and translocation of wild animals can be divided into the following
categories.
•Those used predominantly for restraint.
•Those used for adaptation and reduction of stress.
•Those used for the reversal or reduction of drug effects.
The groups and classes of drugs used for the chemical restraint are summarized in Table 1. Drugs used for the reduction
of stress and adaptation during translocation include long-acting tranquillizers, serotonin antagonists, and appetite
stimulants. Specific antidotes (for opioids, alpha-2-agonists, benzodiazepines and competitive peripheral muscle
relaxants) are used to reverse the depressant effects of drugs.
Adrenergic, antimuscarinic, and analeptic drugs are used either to stimulate central nervous, cardiovascular, or
respiratory functions, or to reduce the side-effects of immobilizing drugs during restraint.
Each group of drugs and the major drugs within each group are discussed separately with respect to their mechanism of
action, chemical and physical properties, pharmacological effects, indications for use, side- and toxic effects, precautions
and contra-indications, antidotes (where applicable), and handling and storage procedures. Suppliers are mentioned
where appropriate.
2 General anaesthetic agents
General anaesthetics are substances that result in loss of consciousness with loss of pain sensation after administration to an
individual. They act pre- and/or post-synaptically, and affect excitatory and inhibitory synaptic transmission in the central nervous
system. Effects on the excitatory synapses are chiefly depressant, whereas they may either depress or enhance inhibitory synaptic
transmission. The actions of anaesthetics are non-specific: they act on a variety of cell types and functions. Deformation of
biomembranes through complex physicochemical mechanisms of expansion, volume change, and fluidization is generally accepted
as the most likely mode of action35.
General anaesthetic drugs are divided into two main groups according to their method of administration viz. inhalation and
injectable anaesthetics. Inhalation anaesthetics (such as halothane, isoflurane and enflurane) are not generally used in wild animals.
They are occasionally used for research and, in the case of captive wild animals, to perform lengthy surgical procedures that require
maintenance anaesthesia. The injectable cyclohexylamines (ketamine, phencyclidine, and tiletamine) and an
alphaxalone-alphadolone steroid anaesthetic mixture are, on the other hand, very important drugs in the wildlife field, being used
particularly in free-ranging and captive wild carnivores. Barbiturates (pentobarbitone and thiopentone) are injectable anaesthetics
that are used only occasionally.
All general anaesthetics, except phencyclidine, tiletamine, and higher concentrations of pentobarbitone, are Schedule 5 substances.
Tiletamine and higher concentrations of pentobarbitone are Schedule 6. These drugs may only be used by a veterinarian, or on
prescription from a veterinarian, in animals. It is an offence to have Schedule 5 or higher schedule medicines in your possession
without a valid prescription from a medical practitioner, dentist, veterinarian, or any other person entitled to issue a prescription. A
permit from the MCC for the importation and use of unregistered Schedule 5 and 6 drugs is required. The only major difference in
control between these two Schedules is that in the case of Schedule 6 drugs a prescription is only valid for 30 days from the day of
issue; a Schedule 5 prescription is valid for six months. Phencyclidine is a prohibited substance, and can only be obtained and used
on a special permit (refer to the legal control section for more details).
2.1 Cyclohexylamines
Cyclohexylamines are a group of drugs that produce a cataleptoid state of immobility, referred to as dissociative
anaesthesia, which is accompanied by marked somatic analgesia35. Spontaneous, unprovoked movements may
occasionally occur in deeply anaesthetized subjects. Muscle tremors and aimless, tonic-clonic movements of the limbs
have also been described. Such movements do not reflect the stage of anaesthesia. Several reflexes that are abolished by
other general anaesthetics are retained, including pharyngeal and laryngeal reflexes. Palpebral and corneal reflexes are
also normally present, and are suppressed only at deep levels of anaesthesia. Other ocular effects such as mydriasis,
nystagmus (involving movement in a diagonal or elliptical path), and eyelids that remain open are characteristic of
dissociative anaesthesia36. Specific licking motions, called the 'serpentine tongue sign', are also characteristic of the
cyclohexylamines23. The sleep component of these drugs appears to be superficial: signs of recovery are indistinct when
consciousness returns, with the animal occasionally appearing to be in a trance-like state. There is persistence of a mesal
sensitivity of certain parts of the skin (such as the scrotum) and of arteries to clamping. Anaesthetic depth is therefore not
assessed by standard evaluation of disappearance of reflexes, ocular effects, and respiratory pattern, but is indicated
mainly by the animal's response to handling and painful stimuli37.
The actual mechanism of action of the cyclohexylamines is not clear, but may be related to increased release of dopamine
in the brain, imbalance of muscarinic-nicotinic cholinergic activity, serotonin metabolism, gamma-aminobutyric acid
(GABA) agonistic effects, or opioid receptor stimulation35,38. The anaesthetic action of these drugs requires a functional
cerebral cortex.
Ketamine, tiletamine, and phencyclidine are currently the only cyclohexylamines of veterinary interest. All three have
been extensively used in a wide variety of mammalian, avian, and reptilian species. They are seldom used alone but are
mostly combined with sedatives in an attempt to overcome their cataleptoid or convulsive characteristics.
Cyclohexylamines are usually administered either intravenously or intramuscularly, but may also be administered orally
under certain circumstances.
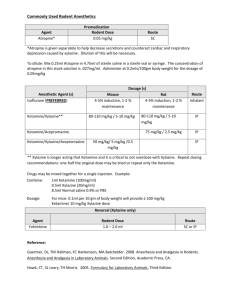
2.1.1 Ketamine
Ketamine hydrochloride ((±)-2-(o-chlorophenyl)-2-methylamino cyclohexanone hydrochloride) is available in
South Africa as Anaket-V® (Twins Pharmaceuticals Ltd) as a 100 mg/ml injectable solution and from
Parke-Davis (Ketalar®) in injectable solution concentrations of either 10, 50, or 100 mg ketamine base per ml.
Anaket-V® is registered for use in dogs and cats, and is supplied in 10 ml multidose vials by Milborrow, the
animal division of Twins Pharmaceutica. Ketalar® is obtainable in vials of 20 ml containing 10 mg/ml and 10
ml for the higher concentrations, but is not registered for animal use. Unregistered ketamine powder can be
obtained on veterinary prescription from Warner Lambert and Kyron Laboratories if higher concentrations are
required.
Chemical and physical characteristics. Ketamine hydrochloride is a white, crystalline powder that is soluble
in water up to a concentration of 20% or more38. The solutions are slightly acidic, with a pH of 3,5-5,5. In the
case of the 10 mg/ml concentration, the solution has been made isotonic with sodium chloride. Benzethonium
chloride at 0,1 mg/ml is included as a preservative in the solutions. Ketamine is a racemic mixture of two
enantiomers: laevo-ketamine (l-isomer) and dextro-ketamine (d-isomer)39,40. Studies have shown that the
d-isomer has three times the anaesthetic potency of the l-isomer. Ketamine and barbiturates should not be
mixed in the same syringe, as they are chemically incompatible and will precipitate. Ketamine is
pharmacologically compatible with other anaesthetics, hypnotics, sedatives, and tranquillizers.
Pharmacological action. Ketamine is a short-acting analogue of phencyclidine, with a relative potency of
approximately 1/5 to 1/6 of that of the parent compound and pharmacological effects typical of the
cyclohexylamine group. Respiratory and cardiovascular functions are generally well maintained. Parenteral
injections take effect within 3-5 minutes, and complete immobilization is produced within 5-10 minutes. The
duration of effect varies with the species and the dose administered. An animal is usually ambulatory within 1-2
hours; however, periods of up to five hours have been reported.
Ketamine is rapidly distributed into all body tissues, primarily adipose tissue, liver, lung and brain. The
recovery from the anaesthetic effects is due mainly to the redistribution of the drug from the brain to other
peripheral tissues. Elimination occurs by biotransformation and formation of water soluble glucoronide
derivatives that are excreted in the urine35. Differences in the elimination pathways between species occur: the
liver appears to be important in the dog, horse, and human, while a large percentage of ketamine is excreted
unchanged by the kidney in rats and cats41. The elimination half-life of ketamine is 66,9±24,1 minutes: this
may be prolonged by the concurrent use of sedatives, e.g., diazepam. Repeated administration of ketamine does
not lead to tolerance of any significance35, although induced hepatic microsomal enzyme systems may
increase the rate of metabolism of the drug.
Indications. Ketamine is used for the immobilization and capture of wild animals. Although it has been used in
a wide range of wild and domestic species23,24, it is particularly indicated in wild carnivores, subhuman
primates, reptiles, and birds9. Due to its relative lack of potency it is used especially in smaller animals24. The
use of ketamine solutions at a concentration of 200 mg/ml has, however, facilitated the successful capture of
larger animals.
In zoo situations where predation is not a problem, ketamine, especially in combination with xylazine, has been
reported as being highly satisfactory for use in ruminants42,43. In free-ranging ungulates, prolonged recovery
and ataxia have been reported42.
Dosage and directions for use. A very large variation in dose of 2-50 mg/kg occurs between species, although
most wild animals require only 10-20 mg/kg. Differentiation occurs between doses required for chemical
restraint with or without total analgesia: intramuscular doses of ketamine of less than 22 mg/kg produce basic
chemical restraint without total analgesia, whereas doses of 22-44 mg/kg produce typical cataleptoid
anaesthesia35.
Ketamine hydrochloride solutions may be administered orally or parenterally. Absorption of ketamine
solutions and immobilization have been achieved by squirting the solution into the mouths of captive cheetahs.
The drug may be administered alone, but is more commonly used together with either xylazine, medetomidine,
diazepam, or azaperone, depending on the species involved. These drugs are combined with ketamine to reduce
its cataleptic and convulsogenic effects. Lower doses are generally required when used in combination with
sedatives and neuroleptics.
Side-effects and special precautions. Many animals experience transitory pain upon injection. The low pH of
the aqueous solutions probably partly accounts for the irritant properties44.
Ketamine produces cataleptoid anaesthetic side-effects typical of the cyclohexylamines. Tonic-clonic
convulsions may occur in a small percentage of wild felids and other carnivores, but these are not as severe as
in the case of phencyclidine. Primates are less commonly affected. Slight central nervous system depression for
24 hours may occur in some felids. Excessive salivation is particularly prevalent in felids: although not
necessary, this can be controlled using atropine sulphate. Since the swallowing reflex is retained, salivation
does not pose a major problem. Furthermore, the use of atropine could cause a serious tachycardia when used
with ketamine.
The eyes of animals anaesthetized with ketamine normally remain open, with the pupils dilated. It is therefore
advisable to use a bland ophthalmic ointment to protect the cornea from desiccation. The eyes should always be
protected from direct sunlight.
The effect on blood pressure varies between species, e.g., in the dog and in man blood pressure is elevated,
while in the rhesus monkey blood pressure is depressed. Ketamine should be administered with caution to
animals with known heart disease. Prolonged apnoea is sometimes observed in large felids. Respiratory
depression and/or paralysis may be exacerbated following the use of muscle relaxants. Precautions should be
taken to control haemorrhage after surgery due to an increase in blood pressure.
Hyperthermia frequently occurs with ketamine immobilization as a result of the catatonic effects of the drug.
This may be aggravated by seizures.
Reversal of drug effects. The alpha-2-adrenoceptor antagonists yohimbine (Yohimbine®, Twins
Pharmaceuticals) at 0,125 mg/kg and tolazoline (Priscoline®, Ciba Geigy) at 0,5 mg/kg have been used as
antidotes for ketamine9,45,46,47. Doxapram (Dopram®, Continental Ethicals) at 1,0-2,5 mg/kg has also been
used successfully to stimulate respiratory depression and reverse anaesthesia following ketamine
immobilization.
Toxicity and particulars of its treatment. Ketamine has a wide safety margin: up to 10 times the usual dose is
normally required for toxicity. Respiratory depression may occur following toxicity. Supportive ventilation
and administration of doxapram are indicated in such cases.
Myoclonic jerking and mild tonic convulsions can be controlled by using ultra-short-acting barbiturates,
diazepam, or midazolam.
Stability and storage instructions. Ketamine solutions must be stored in a cool, dry place below 25°C. Protect
from light. Solutions are stable, and retain their potency over periods exceeding one year.
2.1.2 Tiletamine
Tiletamine hydrochloride (2-(ethylamino)-2-(2-thienyl) cyclohexanone hydrochloride) is an analogue of
ketamine. It is available as a 1:1 combination with Zolazepamzolazepam (a pyrazolodiazepine) known as
Zoletil®. Zoletil® is registered for use in domestic and wild animals in South Africa. It is supplied as Zoletil®
20, Zoletil® 50 and Zoletil® 100 by Palmvet in glass vials of 50 mg, 125, mg and 250 mg tiletamine powder
respectively, in combination with equal amounts of zolazepam powder. The powder is dissolved in a Zoletil®
solvent for use as an injectable solution. The drug is presented in single packs containing one vial of either
Zoletil® 20, 50, or 100 and a vial of solvent.
Chemical and physical characteristics. Zoletil® is a white to yellow lyophilized powder containing tiletamine
and zolazepam in a 1:1 ratio. Zoletil® solvent is sterile water used for reconstitution of the Zoletil® powder.
Pharmacological action. Tiletamine is three to four times more potent than its ketamine congener.
Administered alone, tiletamine produces similar pharmacological characteristics to those of ketamine, although
the duration of action is three times longer35. However, tiletamine does show a proclivity for convulsions
similar to phencyclidine. Zolazepam, when combined with tiletamine, produces potentiation of the anaesthetic
effects of tiletamine, muscle relaxation, abolition of convulsions, and smoother recovery from anaesthesia.
Indications. Zoletil® has been used extensively in a variety of species37,48,49,50,51,52. It is indicated for the
chemical immobilization and anaesthesia of wild and exotic primates and carnivores.
Dosage and directions for use. Dosages of this preparation are expressed in mg of the drug combination.
Recommended dose rates vary widely between species, e.g., in wild carnivores doses were found to range from
1,4-30,0 mg/kg37. The dose rates recommended by the supplier of Zoletil® are: Primates, 5-10 mg/kg;
Ursidae, 3-5 mg/kg; Felidae, 4-5 mg/kg for lions and 4,0-6,5 mg/kg for leopards; and Canidae, 4-5 mg/kg. A
maintenance dose of one third to one half of the initial dose can be given if necessary. Captive, untamed wild
dogs (Lycaon pictus) were successfully immobilized at dosages ranging from 2,3-32,3 mg/kg52.
Depending on the dose required, Zoletil® 20, 50, or 100 powder is reconstituted in the vial with 5 ml of the
solvent to give a final total tiletamine and zolazepam concentration of 20, 50, or 100 mg/ml, respectively. More
concentrated solutions of up to 500 mg/ml can be made by using less of the solvent.
Side-effects and special precautions. Similar to ketamine, although the cataleptoid side-effects of tiletamine
have been reduced by the addition of the diazepinone, zolazepam. The product is contra-indicated in subjects
under systemic organophoshate treatment, and in conjunction with phenothiazine tranquillizers (e.g.,
acepromazine) because of potentiation of the cardiodepressant and poikilothermic effects.
The dose of Zoletil® should be reduced in geriatric and debilitated animals. Noises and bright light should be
avoided during recovery stages. Body temperature should be monitored and animals should be cooled down if
necessary or protected from heat loss during anaesthesia. The eyes remain open with the pupils dilated. The
eyes should be protected from direct sunlight, and a bland ophthalmic ointment should be instilled to protect the
corneas from desiccation.
Reversal of drug effects. Similar to ketamine. The tiletamine effects wear off more rapidly than the sedative
effects of zolazepam. The effects of the latter could be reversed by flumazenil (Anexate®, Roche), a specific
diazepinone antagonist. A total dose of 0,3-1,0 mg of flumazenil has been recommended in humans.
Toxicity and particulars of its treatment. Respiratory depression is the main toxic effect of Zoletil®.
Treatment is symptomatic and supportive.
Stability and storage instructions. Zoletil® must be stored at 15-25°C, and should be protected from light.
Once reconstituted, the solution should be used within 24 hours. The reconstituted solution can, however, be
stored at 4°C and protected from light for several days.
2.1.3 Phencyclidine
Phencyclidine hydrochloride (1-(1-phenylcyclohexyl)piperidine hydrochloride) is listed as a prohibited
Schedule 8 substance in South Africa. A special permit approved by the Director General, Department of
National Health and Population Development, is required for its use. Previously available in a 100 mg/ml
injectable solution as Syclan® injection and Sernylan®, phencyclidine is no longer available in South Africa,
and must therefore be imported.
Phencyclidine is currently only recommended for the immobilization of larger sized lions and primates, and for
hyaenas. It is recommended at a dose rate of 1 mg/kg and lower in primates, 0,7 to 1,5 mg/kg in lions, and
0,7-1,0 mg/kg in hyaenas. Leopards, lions, and hyaenas have been captured by using meat impregnated
phencyclidine as a bait. Chimpanzees have also been anaesthetized with phencyclidine orally by mixing the
drug into their feed73. Oral administration is less predictable in action, and higher doses are necessary.
The pharmacological effects, side- and toxic effects, and the reversal of effects, are similar to those described
for the other cyclohexylamines.
2.2 Steroid anaesthetics
The anaesthetic property of steroids, including progesterone, was originally observed in 194136. Further development led
to steroidal anaesthetics devoid of hormonal activity. A steroid injectable anaesthetic combination,
alphaxalone/alphadolone, is currently the only drug available for use from this group. Minoxolone, a recently developed
steroid anaesthetic, has been evaluated clinically in dogs, but is not yet available for general use36. Minoxolone is
formulated without a surfactant, and therefore avoids the hypersensitivity reactions associated with
alphaxolone/alphadolone, which contains the surfactant Cremophor EL.
2.2.1 Alphaxalone/alphadolone
Alphaxalone and alphadolone acetate are two pregnanediones that are mixed and solubilized in saline with 20%
w/v polyoxyethylated castor oil into the final product. The product is available as Saffan® from Janssen
Pharmaceutica as an injectable solution of 12 mg/ml of total steroids, composed of 9 mg alphaxolone and 3 mg
of alphadolone acetate. It is supplied in packages of either 10x5 ml or 10x10 ml glass ampoules.
Chemical and physical characteristics. Alphaxalone is water insoluble, and attempts to form water-soluble
salts produced molecules with a high degree of CNS stimulant activity36. Alphadolone acetate used in
combination with alphaxalone acetate increases its solubility three-fold. Further solubilization into an aqueous
formulation is achieved by the addition of 20% of the non-ionic surfactant polyoxyethylated castor oil
(Cremophor EL). The final formulation is a slightly viscous, sterile, clear solution, that has a neutral pH.
Pharmacological action. The steroid mixture produces short-acting anaesthesia with good muscle relaxation.
Respiration is usually well maintained. Alphadolone is less potent than alphaxalone. Induction of anaesthesia
following intravenous administration is rapid and smooth. The duration of anaesthesia is short: within 6-9
minutes following a single intravenous injection of the product, animals are able to stand and, after a period of
ataxia, a rapid return to normal is observed. Complete recovery following intramuscular administration takes
slightly longer. However, the depth and duration of anaesthesia remains dose dependent. Surgical anaesthesia
is achieved for 5-10 minutes at higher doses. Anaesthesia can be maintained for prolonged periods with
incremental doses, or by continuous intravenous infusion, without prolonging post-anaesthetic recovery.
The steroid components are biotransformed in the liver to polar metabolites that are excreted in the bile. The
plasma half-life of alphaxalone in various animals is approximately seven minutes. About 60-80% of
radio-labelled compounds are excreted in the bile within the first three hours after administration of the product
in rats36. Accumulation does not occur following repeated administration. Termination of effect is not
dependent on uptake by adipose tissue.
The hormonal effects of alphaxalone and alphadolone are less than one-sixtieth of those of betamethasone, and
are slightly less than those of hydrocortisone35. There is no known mineralo-corticoid or progestational
activity. Very weak anti-oestrogenic activity does occur: Saffan® is capable of antagonizing the uterotropic
action of exogenous oestradiol.
Indications. Saffan® is registered as an injectable steroid anaesthetic for use in cats and monkeys. It has also
been used for the immobilization and sedation of birds and reptiles35. Its main indication in wild animals,
however, has been the immobilization of cheetahs54.
Saffan® is contra-indicated in dogs because the solubilizing castor oil derivative causes the release of
histamine or histamine-like substances in this species. These effects can be avoided by the concurrent use of
antihistamines such as promethazine (Phenergan®, Maybaker).
Dosage and directions for use. An intravenous dose of 2,2-3,3 mg/kg in cheetah and 6-9 mg/kg in monkeys is
recommended. Higher doses are required when administered intramuscularly, e.g., 12-18 mg/kg in monkeys
for surgical anaesthesia. Saffan® must be given by deep intramuscular injection to ensure consistent results.
Anaesthetic effects can be maintained for prolonged periods by repeated intravenous administration or
continuous infusion.
Saffan® is compatible with commonly used volatile and gaseous anaesthetic drugs and premedicant drugs, but
profound hypotension has been recorded when it is injected intravenously after barbiturates and
alpha-chloralose.
Side-effects and special precautions. Alphaxalone-alphadolone has a wide safety margin in laboratory animals
as well as in animals during clinical use. This is probably due to minimal depression of respiration in
comparison to most other injectable anaesthetics. The net effect on cardiovascular function is hypotension.
Sinus rhythm was recorded in all cheetahs examined, and a rapid heart rate was present54. Saffan® also
induces a state of hypothermia in cheetahs, and should be used with caution on hot and cold days55.
In cats, sneezing on induction, flushing, and unilateral or bilateral oedema of the paws, ears, and face are
sometimes seen. These effects are not of clinical significance, and usually resolve within an hour. Salivation
may occur in the monkey. Very occasionally cats may experience respiratory embarrassment shortly after
induction. This may involve histamine release, but the true underlying cause is unclear. Obstruction of the
airway, fear, and pre-existing lung pathology affecting lung compliance under anaesthesia have been identified
as possible predisposing causes. In these cases, immediate treatment should be given: an adequate airway
should be established and oxygen enriched positive pressure ventilation commenced. Intravenous
corticosteroids and respiratory stimulants, such as aminophylline, should be given. The use of antihistamines
should also be considered. Such incidents can be minimized by slow intravenous injection of Saffan®,
intubation with care for inhalation anaesthesia, the use of neuroleptics or sedatives to sedate anxious cats, and
treatment of chronic infections before the anaesthetic is given. Another complication in cats is a rare occurrence
of necrotic lesions on the extremities 8-10 days following anaesthesia with Saffan®. The cause is unknown.
The release of histamine and histamine-like substance caused by the castor oil derivative in dogs results in
severe vasodilation and hypotension. Symptoms include generalized erythema, oedema (especially of the face),
marked hypotension, tachycardia, intense bronchospasm, and abdominal pain. Immediate anaphyllactic
treatment is required.
Reversal of drug effects. There is no known antidote available.
Toxicity and particulars of its treatment. Toxicity is manifested as prolonged anaesthesia, respiratory
depression, coma, and death. Administer respiratory stimulants such as doxapram (Dopram®, Continental
Ethicals), intravenous fluids, oxygen and generalized shock treatment.
Stability and storage instructions. Saffan® is stable if stored at room temperature.
2.3 Barbiturates
ThiopentonePentobarbitonePentobarbitone (Sagatal®, Maybaker Animal Health) and thiopentone (Intraval Sodium®,
Maybaker Animal Health) are very seldom used in wild animals. In the past, they have been used to treat the epileptiform
convulsions induced by the cyclohexylamines. They have also been used to prolong anaesthesia in captive wild animals
following immobilization. Higher concentrations of pentobarbitone (Euthatal®, Maybaker Animal Health; Euthanase®,
Milborrow Animal Health) are used for euthanasia of small animals.
Thiopentone may only be administered intravenously: extravascular administration is severely irritant, resulting in
perivascular necrosis. Pentobarbitone is also administered intravenously, but may be administered intramuscularly or
intraperitoneally. Lions fed meat from horses euthanatized with pentobarbital have become anaesthetized.
2.4 Inhalation anaesthetics
Inhalation anaesthetics are not generally used for the immobilization of wild animals. However, veterinarians are
experiencing a growing need for prolonged and controlled anaesthesia to perform complicated surgery or meticulous
diagnostic procedures9. Inhalation anaesthetics are also used to induce anaesthesia in animals that can be manually
restrained, such as birds, reptiles, and young mammals. They are otherwise used to augment analgesia, or to increase the
depth of anaesthesia, in animals initially immobilized using injectable agents.
Inhalation anaesthetics have the advantage that the level of anaesthesia can be controlled precisely during prolonged
surgical procedures. Uptake and removal of the drug are very rapid through the respiratory system. An endotracheal tube,
which is required for inhalation anaesthesia using semi-closed and closed circuit systems, is also a means for support of
pulmonary ventilation should this become necessary.
Inhalation anaesthetics are divided into volatile and gaseous substances. Halothane (Fluothane®), methoxyflurane
(Pentrane®), enflurane (Ethrane®) and isoflurane (Forane®) are the volatile drugs, and nitrous oxide the gas, that are
available. Nitrous oxide is never given alone, but is used in combination with the other inhalation anaesthetics, such as
halothane, to reduce the dose required. It has a potent inherent analgesic effect. Isoflurane and enflurane have the
advantage that they are less arrhythmogenic than halothane and methoxyflurane. The latter drugs sensitize the
myocardium to the effects of catecholamines such as adrenaline and nor-adrenaline. Inhalation anaesthetics can be
administered by auto-inhalation, but this is not recommended in wild animals: they are best administered with an
anaesthetic machine in a closed or semi-closed configuration. Specific anaesthetic machines are required for all volatile
anaesthetic drugs: only a few can be used interchangeably. Dose-dependent cardiac and respiratory depression are the
main side-effects of these drugs.
3 Opioid analgesics
Opioid is a term used to designate a group of drugs that are, to varying degrees, opium- or morphine-like in their properties.
Previously, these drugs were referred to as narcotics. Since the term in a legal context now refers to any drug that can cause
dependence it is no longer useful in a pharmacological context56. Opiates are drugs derived from opium, such as morphine,
codeine, and the many semi-synthetic congeners of morphine, e.g., etorphine. Entirely synthetic substances with morphine-like
activity are referred to as opiate or morphine substitutes.
Opioids interact with several closely related groups of receptors (mu, kappa, delta and sigma)56,57 collectively known as the
endorphin receptors. These receptors occur in the brain, spinal cord, autonomic nervous system, myenteric plexus of the
gastrointestinal (GI) tract, heart and kidney, as well as in other organs35. They are important in neuromodulation of pain, moods,
emotional response, behaviour, voluntary muscle tone, and GI motility. The occurrence and distribution patterns of the various
endorphin receptors in the body, and the consequences of their activation, may vary significantly within an animal and between
species58. It is also possible that receptor population changes could occur as result of external factors (such as when animals are
kept in captivity), and as a consequence of stress.
The effect of opioid drugs on the endorphin receptors differs in terms of affinity and intrinsic activity. A given opioid drug may
interact to a variable extent with all or some of the endorphin receptors, and may act as either an agonist, as a partial agonist, or as
an antagonist. It is also possible that a drug may be agonistic on certain receptors, but only partially agonistic or even antagonistic
on others56. A great variation in the pharmacological effects of opioid drugs, both between species and under different
physiological conditions, can therefore be expected.
Opioids have a broad range of pharmacological effects35. They are extremely potent analgesic drugs, and have variable
psychotropic effects in different animal species. For example, CNS depression is observed in the dog, monkey, human, and in many
wild animal species, while in feline, porcine, and equine species excitatory effects are observed. The reasons for these differences
are not well understood: they have been ascribed to a number of factors, including alteration in the functioning of the brain
dopaminergic or noradrenergic systems59, and to differences in the distribution pattern of the opiate binding sites in the brain58.
Excitatory effects, including convulsions and extrapyramidal signs such as myoclonic twitches, catalepsy, and muscular rigidity,
have largely been attributed to overdosage35. Respiratory depression, hypertension or occasionally hypotension, inhibition of the
thermoregulatory mechanism, inhibition of gastro-intestinal motility, and other important effects need to be considered when using
these drugs.
Opioids, being dangerous, habit-forming drugs, are listed under Schedule 7. As such, the veterinarian and pharmacist shall keep a
register or permanent record of the following:
•The quantity of any such drugs possessed, imported or acquired;
•The date of acquisition;
•The name of the person from whom and the place from which the same were imported or acquired; and,
•The quantity that has been disposed of and whether this was by sale or by use in the ordinary course of practice.
Every register shall be retained and preserved for at least three years, and must be balanced every three months on the last day of
March, June, September, and December of each year. The register is open to inspection by any person authorized thereto in writing
by the Registrar of Medicines and Related Substances. A prescription for a Schedule 7 drug may not be repeated and is only
valid for 30 days from the day of issue. Furthermore, the amount prescribed may not exceed that which is required by a
person for 30 days.
Currently the most common opioids used for the immobilization of wild animals are etorphine, fentanyl, and carfentanil. Etorphine
is the only opioid registered for use in South Africa. Fentanyl has been exempted from registration under Section 36 of the Act, and
is available directly from the manufacturer. Sufentanil and A3080 are newer opioids currently being investigated for use in wild
animals. Butorphanol (Torbugesic®, Ayerst Laboratories), meperidine (Pethidine®, Glaxo) and diethylthiambutene (Themalon®)
are other opioids occasionally used for the immobilization of captive wild animals60.
Neuroleptanalgesic drugs are combinations of neuroleptics and opioids. Droperidol-fentanyl (Innovar-Vet®, Janssens (Pty) Ltd) is
a registered combination that is used in wild animals.
Opioid drugs have occasionally been dissolved in dimethylsulphoxide (DMSODMSO)23. The rationale behind this
procedure is that high concentrations can be achieved, and the DMSO increases the rate of absorption of the drug in the
injected animal. Unfortunately, absorption following accidental human injection is significantly accelerated. Furthermore,
the DMSO facilitates the absorption of the dissolved drug through the intact skin. As even the small quantities of drug
remaining in the dart and needle are sufficient to kill a human this practice is not recommended, and has largely been
discontinued.
3.1 Morphine derivatives
3.1.1 Etorphine
Etorphine hydrochloride (6,14-endoetheno-7alpha-(2-hydroxy-2-pentyl)-tetrahydro-oripavine hydrochloride)
is a semi-synthetic opioid derived from thebaine. Etorphine is available in South Africa as M99® from
Krüger-Med Pharmaceuticals (Pty.) Ltd. in 4,9 mg/ml and 9,8 mg/ml concentrations. M99® is supplied in
preformed polystyrene containers containing a 10 ml rubber-stoppered vial of M99® and a similar vial of
M5050® (diprenorphine). Both vials contain 5 ml of solution.
Chemical and physical characteristics. Etorphine hydrochloride powder is readily soluble in slightly acidified
water23. Solutions of up to 10 mg/ml may be made in dimethyl sulphoxide (DMSO). The solutions are clear
and colourless.
Pharmacological action. The pharmacological effects of etorphine are typical of the opioids, but may vary
significantly between species. Its analgesic potency is given as 500 to 10000 times that of morphine sulphate,
depending on the nature of the test9,21,24,35,61.
Given alone, etorphine causes areflexia without total loss of consciousness23. The initial reaction of a wild
animal following administration of etorphine varies with the method of approach: this may range from panic
flight with a helicopter to passive squatting in a net or trap24. Changes in gait or walking pattern occur in most
terrestrial species: these progress steadily as the animal loses awareness of obstacles and terrain. In most
ruminants this ataxia is accompanied by the characteristic 'hackney gait'. The more nervous animals, such as the
eland, kudu, and waterbuck, continue to run for extended periods, especially if etorphine is given alone. During
this period, ruminants can often be approached and guided while on their feet. Apart from gait changes, an early
sign of induction in elephants is relaxation of the trunk.
As immobilization progresses, animals either suddenly collapse into sternal or lateral recumbency or stop,
often unimpeded, and sway before going down. The onset of action following standard doses is about 2-4
minutes, up to eight minutes in some individuals23,24. Peak effects differ according to the rate of absorption
but usually follow in 15-30 minutes, the duration being about one hour. The drug is excreted mainly through the
liver and intestine. Re-cycling of etorphine, either from enterohepatic circulation or redistribution from adipose
depots, occurs, occasionally leading to Renarcotizationrenarcotization of the animal23,24.
Indications. Etorphine is used for the immobilization and capture of all African ungulates, but in particular for
the very large ungulates and sub-ungulates such as elephant, rhinoceros, giraffe, zebra, wildebeest, and
hippopotamus (when on land)23. It is generally not suitable for use in carnivores, but has been used in spotted
hyaenas62 and bears.
Dosage and directions for use. The dosage rate of etorphine varies from 0,5 µg/kg in the rhinoceros to 5 µg/kg
in camels35. The recommended total doses for immobilizing healthy adults of some of the common game
animals in southern Africa63 are given in Table 2. Young, old, sick, or debilitated individuals generally need
lower doses than those recommended for adults. Females of any species also require a slightly lower dose
relative to males. Pregnant animals appear to be more resistant to the effects of etorphine than non-pregnant
ones63. It is important to emphasize the difference in sensitivity of free-living animals, captive wild animals,
and domestic animals to immobilizing drugs24: tamer animals generally require much lower doses.
In many cases etorphine is administered alone. In some antelope species and rhinoceros it may be combined
with either a sedative (e.g., xylazine), a tranquillizer (e.g., acetylpromazine or azaperone), or an antimuscarinic
(e.g., hyoscine) to shorten the knock-down time and some of the adverse reactions in these species.
Side-effects and special precautions. The most important side-effects are respiratory depression, rumen stasis
and a decrease in gastro-intestinal motility, changes in heart rate and blood pressure, decrease in urine output,
and excitement. Of these, excitement has caused most problems, but has been overcome by the judicious use of
sedatives or tranquillizers23. Depression of respiratory rate and minute volume29 may result in a severe
hypoxia, hypercapnoea, and progressive acidosis35. Alterations in cardiovascular function have been fatal in
equids64. Hypertension has been reported in a number of domestic ungulates65, and presumed in non-domestic
ungulates based on cardiovascular responses66. Recently, hypertension following etorphine administration in
rhinoceros has been reported65. Passive regurgitation of rumen contents, which may result in fatal aspiration, is
not uncommon9,35.
Reversal of drug effects. Spontaneous recovery from etorphine-induced immobilization is slow9. Opiate
antidotes, including diprenorphine, nalorphine, and naloxone, have been used with success to reverse the
depressant effects of etorphine. The recommended total doses of diprenorphine and nalorphine following
etorphine administration in wild animals are given in Table 2. Diprenorphine (M5050®) is the registered
antidote for etorphine in South Africa. It is recommended at 3-5 times the M99® dose in elephants, rhinos, and
giraffes, and at 2-3 times the M99® dose in other species. Most frequently it is used at between 2:1 and 1,2:1
diprenorphine to etorphine ratio (w/w).
Naloxone is a pure opioid antagonist, in contrast to the other previously mentioned antidotes which have both
antagonistic and some agonistic properties35. Naloxone has a short half-life, and high doses are required to
prevent renarcotization. This drug is of particular importance in emergency situations (due to its rapid effect).
The low concentrations (0,4 mg/ml) of the naloxone formulation available limits its use in the wildlife field.
Naloxone has a short half-life, and high doses are required to prevent renarcotization. The effects are dose
dependent, and larger species may require two injections because of the short half-life24. The first dose is
administered subcutaneously at 0,04-0,07 mg/kg followed by an intravenous or occasionally an intramuscular
injection at 0,1-0,25 mg/kg.
Toxicity and particulars of its treatment. Toxic effects of opioids are principally related to respiratory
depression. Certain species or individuals may be particularly sensitive24. No supportive treatment is required
under normal conditions23. Overdosage is readily treated with an appropriate antagonist and, if necessary, with
analeptics, e.g., doxapram (Dopram®, Continental Ethicals).
Humans are particularly sensitive to the toxic effects of etorphine. The rapid onset of toxic effects makes it
extremely dangerous, the effects being evident within two minutes of injection and reaching a peak in 5-10
minutes23. Symptoms and signs of a toxic dose include loss of consciousness, slow, shallow respiration,
cyanosis, pin-point pupils, weak pulse and fall in blood pressure, and twitching of the muscles. Death occurs in
deep coma by respiratory failure.
In the event of accidental poisoning, 1 ml of naloxone 0,4 mg/ml (Narcan®, Boots) must be administered
immediately IM or preferably IV. Repeat at 2-3 minute intervals until symptoms are reversed. Any one of the
other antidotes could also be used, but only if naloxone is not available. Care should be taken due to possible
aggravation of respiratory depression by these mixed antagonists. Additional first aid treatment: see
Prevention and Management of Capture Drug Accidents, this Section.
Stability and storage instructions. M99® solutions are stable if stored in a cool place (below 25°C) away from
light. Keep the drugs in the container supplied.
3.2 Opiate substitutes
3.2.1 Fentanyl
Fentanyl citrate (1-(2-phenethyl-4-(N-propionyl-anilino))-piperidine) (Sublimaze®) is not readily available in
South Africa, and is usually only obtainable directly from the manufacturer, Janssen Pharmaceutica, on
request. It is supplied as fentanyl citrate powder in 0,1 g and 1,0 g sachets. A droperidol-fentanyl combination
solution (Innovar Vet®) is readily available from Janssen Pharmaceutica.
Chemical and physical characteristics. Fentanyl is a pethidine analogue. The citrate salt of fentanyl is a white,
crystalline powder. One gram of the citrate salt contains 0,637 g of fentanyl base. It is sparingly soluble in
water, giving solutions with a slightly acid reaction (pH 3,8-5,0). Solutions can be prepared for injection in
concentrations of up to 30 mg/ml67. More concentrated solutions may be prepared in other solvents: solutions
containing 20 mg and 40 mg per ml respectively have been prepared by dissolving 1,57 g and 3,14 g fentanyl
citrate in dimethyl sulphoxide (DMSO) to make 50 ml23.
Pharmacological action. Similar to the opiates, with an analgesic potency about 100 times that of
morphine23,24. One of the principal advantages of fentanyl is that many species (e.g., blesbok, hartebeest)
remain standing and can be worked on in this state. It also appears to have less effect on the respiratory centre.
Fentanyl has a rapid onset of action and short duration of effect. The onset of effect occurs between 3-5
minutes, with a peak reached in about 15 minutes23. The peak effect lasts for approximately 30 minutes. It is
rapidly metabolized in the liver, and its metabolites are excreted in the urine. The short duration of action is
probably due to redistribution rather than metabolism and excretion68.
Indications. Fentanyl is effective for immobilizing most of the African antelope as well as buffalo, giraffe,
hippopotamus, rhinoceros, and warthog23. Zebras are fentanyl-resistant, and cannot be captured with this
drug63. In most instances it is used in combination with either azaperone, xylazine, or hyoscine.
Azaperone-fentanyl immobilized animals tend to remain standing. The droperidol-fentanyl combination
(Innovar Vet®) has been used in smaller carnivores.
Dosage and directions for use. Recommended total doses of fentanyl for some common wild animals in South
Africa are given in Table 2. Fentanyl will immobilize white rhinos but, unless very large doses are used, they
remain sensitive to noise and are inclined to get up and move off if disturbed69.
Pharmacological solutions containing 10 mg/ml of the free base may be prepared by dissolving 0,785 g of
fentanyl citrate in distilled water to make 50 ml. The water is slightly warmed before dissolving. Sterilization of
solutions, if needed, is performed by filtration. A bacteriostatic, such as methyl- or propyl-paraben, may be
added by dissolving in nearly boiling water before the products are added. Solutions of fentanyl are best made
up by a pharmacist63.
Side-effects and special precautions. Similar to etorphine, but with a lower occurrence. Side-effects are
mainly depression of respiration, especially where fentanyl is used alone. Unpredictable bradycardia may occur
in dogs70. Clinically, its use is undesirable in patients where respiration is significantly depressed, and in cases
of advanced liver or kidney disease. It should also be used with caution in combination with central nervous
system depressants, since fentanyl has been shown to potentiate, or to be additive to, compounds such as
sodium pentobarbitone23.
Allotriophagia is a marked feature, and care must be taken that animals do not choke on large quantities of
herbage.
Reversal of drug effects. The action of fentanyl may be reversed with the usual opioid antagonists such as
diprenorphine (M5050®) and nalorphine. Doses are given in Table 2. Supportive treatment consists mainly of
atropine to counteract the bradycardia.
Toxicity and particulars of its treatment. Concentrated solutions of fentanyl in DMSO should be handled with
care because of the danger of absorption through the skin. In the case of accidents, emergency treatment as for
etorphine should be carried out (see Prevention and Management of Capture Drug Accidents, this Section).
Stability and storage instructions. Fentanyl base, solutions, and combinations with azaperone remain stable
for three to four years. Temperatures of up to 40°C do not affect stability.
3.2.2 Carfentanil
Carfentanil is a 4-amino-piperidine derivative, and is the most potent opioid known60. It can be used
successfully at doses as low as 1 µg/kg. Since the first report of the use of carfentanil for a variety of African
wildlife species71 this drug has been used widely for the immobilization of both free-ranging and captive wild
animals60. Burchell's zebras are refractory to carfentanil71. Carfentanil has a wide safety margin, with dosages
ranging from 0,6-28,6 µg/kg. Giraffes, which are usually caught standing or ambulatory, become recumbent
without side-effects. Difficult species such as waterbuck show excitability in the induction phase71. Even at
high doses, respiratory depression is rare.
Carfentanil is not available in South Africa, but can be imported from the USA through Janssen Pharmaceutica.
The drug is supplied as a 3 mg/ml solution of carfentanil citrate in 10 ml bottles. A special import permit is
required from the MCC.
Carfentanil, probably due to its high potency and long duration of activity, has a major disadvantage of
occasionally producing renarcotization following administration of an antagonist. Renarcotization, or
prolonged agonist effects, have resulted in severe metabolic problems and death60.
3.2.3 Sufentanil
Sufentanil (Sufenta®, Janssen Pharmaceutica) is a carfentanil-related compound about half as potent as
carfentanil, but with a higher therapeutic index. It is used in humans and in small animal medicine, and has only
occasionally been used in wild animals. Successful immobilization of gray wolves Canis lupus with sufentanil,
alone and in combination with xylazine, has recently been reported72. A severe bradycardia, and at higher
doses a negative inotropic effect, have been observed73.
3.2.4 A3080
A3080 is a newer opioid agonist that has a significantly shorter duration of action than carfentanil or etorphine
in laboratory animals, and is only slightly less potent than carfentanil74. The drug has been used successfully
for the immobilization of several wild animals74,75. At the time of publication the drug is still only available
for experimental purposes.
4 Hypnotics and sedatives
Hypotnics are drugs used to moderately depress the central nervous system . They produce a physiological state of sleep from
which a subject can easily be aroused. A sedative may be used to quieten an animal that is excited by unfamiliar handling
procedures or by a change in its surroundings. Hypnotic and sedative effects are dose dependent, with only slight sedation at low
doses, increasing sedation with increasing doses, until hypnotic effects are attained. Similarly, increasing intensity of hypnosis is
achieved with still higher doses, until the animal becomes comatosed at excessively high doses. These drugs are seldom used alone
in wild animals, but are extensively used in combination with immobilizing drugs, or immediately following restraint, for their
anticonvulsant, muscle relaxant, or sedative activity. Some members of this group of drugs are able to produce general anaesthetic
effects, and may be used on their own.
All hypnotics and sedatives registered in South Africa are listed as Schedule 5 substances. Refer to the legal section and section on
general anaesthesia for details on control procedures of these substances.
There are two main groups of drugs classified as hypnotics and sedatives (viz. diazepinones and alpha-2-adrenoceptor agonists)
which are used in wild animals (Table 1). Some of the most recently developed drugs (e.g., climazolam and medetomidine) used for
the immobilization of wild animals belong to these groups. These drugs are administered by intravenous or intramuscular injection.
Diazepinones may also be administered orally.
4.1 Diazepinones
The effects of the diazepinone derivatives stem from their affinity for specific receptors in the brain and spinal cord that
have been identified using radio-labelling techniques. Responses after their administration are to a large degree central in
origin. It has been postulated that the diazepinones produce their effects in two ways: by modification of ionophore
porosity in conjunction with gamma-aminobutyric acid (GABA), and by the promotion of the central neuronal inhibition
by glycine76. This is illustrated in Fig. 1. The general pharmacological effects of the diazepinones are sedation,
anxiolysis, anticonvulsion, muscle relaxation, and respiratory depression.
Diazepam, midazolam, climazolam, and zolazepam are members of the diazepinone group that have been used in wild
animals. Zolazepam is available as a tiletamine-zolazepam combination (Zoletil®, Palmvet) and has been discussed
under the general anaesthetics. Climazolam is not available in South Africa, but has been used experimentally in wild
animals in this country. The recent development of a benzodiazepine antagonists, such as flumazenil, holds promise for
more extensive use of the general anaesthetic characteristics of midazolam and climazolam for the immobilization of
wild animals76,77,78.
4.1.1 Diazepam
Diazepam (7-chloro-,-dihydro-1-methyl-5-phenyl-2H-1,4-benzodiazepine-2-one) is available as a 5 mg/ml
injectable solution, as an oral tablet, and as a syrup (Valium®, Roche). The injectable solution is supplied in 5
x 2 ml ampoules. Other similar injectable solutions of diazepam available are Pax® (Labethica) and
Scriptopam® (Propan Generics). None of the diazepam drugs available in South Africa are registered for
veterinary use.
Chemical and physical characteristics. Diazepam is a colourless, crystalline substance that is only slightly
soluble in water, is soluble in alcohol, and is freely soluble in chloroform. The injectable solution has a pH of
6,2-7,0. Diazepam is absorbed by plastics substantially, and should therefore be stored and administered in
glass containers.
Pharmacological action. The pharmacological effects of diazepam are similar to those that have been
described for the diazepinone group. A complex relationship exists between the pharmacokinetics and clinical
efficacy of diazepam36. Differences exist between species in the biological half-life and the number of
biologically active metabolites formed. The proportions of these metabolites formed vary depending on the
route of administration. The rates at which these metabolites penetrate the CNS may also vary. Parenteral
administration of diazepam results in the highest CNS availability of the more active, unchanged drug.
Metabolites, of which N-desmethyl-diazepam (or nordiazepam) is the most important35, have approximately
one third of the activity of the parent compound. The biological half-lives, and the extent and types of
metabolism, are not known for the various wild animal species. This could explain the unpredictability of
clinical efficacy of diazepam in these animals.
Onset of pharmacological effects occurs within 1-2 minutes of intravenous injection, and within 15-30 minutes
when given intramuscularly, depending on the dose9. Clinical effects usually disappear within 60-90 minutes.
Indications. Diazepam is indicated for sedation and anxiolysis of wild animals during transportation, and for
muscle relaxation and as an anticonvulsant following immobilization. The latter applies particularly to the
cyclohexylamine anaesthetics, but also to the opioids. It may be used as premedication before surgery in wild
animals that can be handled manually to facilitate inspection, and also for the treatment of viciousness and
nervous agalactia. Diazepam has been used successfully in a number of zoo animals, including antelope and
zebras. Concurrent intravenous administration of diazepam and ketamine has been used successfully on
various raptors (1,0-1,5 mg/kg and 30-40 mg/kg, respectively), river otters (0,32-0,58 mg/kg and 17-30 mg/kg,
respectively) and seals (0,05-0,25 mg/kg and 1-10 mg/kg, respectively)35. It has also been used as a
premedication at 0,37 mg/kg in alligators immobilized with suxamethonium (Scoline®, Glaxo).
Dosage and directions for use. The dosage varies from 0,5 to 10 mg/kg depending on the species, purpose, and
degree of excitement at the time of injection9,23. Intravenous injections must be injected into a large vessel
such as the jugular, and should be administered slowly.
Diazepam should be used alone, as it is incompatible with most aqueous solutions of other drugs, and
precipitation of the active substance will ensue. Valium® (Roche) can, however, be used in a 5-10% glucose
infusion or in an equal volume of 0,9% saline. The contents of the ampoules (not more than 4 ml) must be
mixed quickly and thoroughly with the total volume of the infusion medium, which should be at least 250 ml,
and the infusion begun immediately.
Side-effects and special precautions. Diazepam is well tolerated in large doses. Respiratory depression occurs
only at higher doses. Paradoxical reactions such as hyper-excitement could occur.
Special precautions must be taken with the intravenous injection of diazepam: too rapid injection, or the use of
veins with too small a lumen, carries the risk of thrombophlebitis. Intra-arterial injection must be avoided
because of the danger of necrosis.
Care should be taken when dosing debilitated or pregnant animals.
Reversal of drug effects. The effects of diazepam can be reversed using either flumazenil (Anexate®, Roche)
or the experimental drugs RO 15-1788 and RO 15-3505. These drugs are specific antagonists of the
diazepinone receptors. Flumazenil is recommended at a total intravenous dose of 0,3-1,0 mg in humans.
Toxicity and particulars of its treatment. The wide safety margin of diazepam reduces the danger of
inadvertent poisoning.
Stability and storage instructions. The product is stable if stored at below 25°C and kept out of light.
4.1.2 Midazolam and climazolam
Midazolam and climazolam are benzodiazepine derivatives used for sedation and induction of
anaesthesia. Climazolam has been used in a variety of wild animals, either alone or in combination with
ketamine, xylazine, or etorphine 77. It is currently not available in South Africa. Midazolam
(Dormicum®, Roche) has recently been registered for use in humans, and is available as 1 and 5 mg/ml
injectable solutions and as a 15 mg tablet. This product has only been used on a few occasions in wild
animals, including the capture of lions fed meat impregnated with midazolam (Morkel, pers. comm.).
These drugs have great potential for wider use in wild animals in the future following the development
of the diazepinone antagonists.
4.2 Alpha-2-adrenoceptor agonists
Alpha-2-adrenoceptors can be found in different locations, both in neuronal and non-neuronal tissues79. At
present, these receptors have been classified into at least three alpha-2-isoreceptors (alpha-2a, 2b & 2c) according
to their affinity for alpha-2-adrenoceptor ligands80,81. These receptors act pre-synaptically (causing feedback
inhibition of noradrenaline release in sympathetic nerve endings), by modulating the function of other neurons
(such as those utilizing acetylcholine, dopamine, and serotonin, reducing the release rate of these
neurotransmitters after neuronal activation), by post-synaptic mediation of constriction of vascular smooth
muscle in the periphery (post synaptic alpha-2-adrenoceptors appear also to exist in the central nervous system),
or on 'hormonal' receptors situated outside the nervous system (e.g., in platelets, pancreas, and adipose tissue)
which are thought to respond primarily to adrenaline in the circulation82.
Alpha-2-adrenoceptors play an important role in modulating sympathetic nerve functions, and in the regulation
of vigilance, cognition, nociception, and cardiovascular function. Pharmacological responses mediated by
alpha-2-adrenoceptors are summarized in Table 3.
These drugs have been used in wide range of non-domestic herbivores and carnivores. They are used mainly as
synergists: mixed with either opioids or cyclohexylamines, they result in reduction of the doses required,
improvements in induction times, and better relaxation. Xylazine was the first drug of this group to be used. It has
recently been followed by detomidine, with medetomidine in the process of registration in South Africa. Specific
alpha-2-adrenoceptor antagonists such as yohimbine, tolazoline, atipamezole, and idazoxan are used to reverse
the effects of these drugs.
4.2.1 Xylazine
Xylazine (2(2,6-dimethXylazineylphenylamino)-4-H-5,6-dihydro-1,3-thiazine hydrochloride) is
available as a 20 mg/ml injectable solution (Rompun® 2%) and as 500 mg powder (Rompun® Dry
Substance) for injection after mixing with a diluent. These drugs are manufactured and supplied by
Bayer Animal Health, South Africa. The injectable solution is sold in 25 ml glass injection vials, and the
powder in a carton containing five multidose glass vials (each containing 500 mg xylazine) and one vial
containing 50 ml of sterile solvent.
Chemical and physical characteristics. Xylazine is an organic base. As xylazine hydrochloride, the
powder is a colourless crystal with a bitter taste. It is readily soluble in water and methanol, is soluble
with difficulty in ether and chloroform, and is practically insoluble in hexane.
Pharmacological action. The pharmacological effects of xylazine conform to those described for
alpha-2-agonists. In addition to alpha-2-adrenergic activity, xylazine also has some alpha-1-adrenergic
effects35 that may be responsible for some of the peripheral vascular effects of xylazine.
Immobilization occurs within 3-5 minutes after intravenous administration, or within 10 to 15 minutes
after intramuscular injection. Analgesia lasts for 15-30 minutes, and hypnotic effects for 1-2 hours.
Painful procedures should not be performed after 30 minutes following administration. Analgesia
appears to be optimum after intravenous administration.
Xylazine undergoes rapid metabolism in the liver, and is excreted in the urine. It has a very short
plasma half-life of 23,0-49,5 minutes in domestic animals35. The duration of many pharmacodynamic
effects of xylazine, such as polyuria, hyperthermia, hypothermia, and diarrhoea, exceed, and are
unrelated to, its plasma half-life.
Sedative potency varies between species, and there is considerable variation in the degree and location
of analgesia, the latter being minimal at the extremities1. Xylazine strongly potentiates the effects of
tranquillizers, sedatives, opioids, and dissociative and other anaesthetic agents83.
Indications. Xylazine is used for sedation, immobilization, mild to moderate analgesia, and muscle
relaxation. It can be used alone at relatively high doses to induce immobilization or, more commonly, as
a synergist in combination with cyclohexylamine anaesthetics or opioids1. The use of xylazine alone or
in various combinations has been established for carnivores, ruminants, equids, and other mammals.
Dosage and directions for use. Rompun® injectable solution (containing 20 mg/ml xylazine) or
Rompun® Dry Substance can be used. Rompun® Dry Substance contains 500 mg xylazine powder that
is dissolved in the diluent supplied. It can be diluted to make up various concentrations, such as 50, 100,
200 or even 500 mg/ml by dissolving the powder in 10, 5, 2,5 or 1 ml of solvent, respectively.
The dose requirements for various species differ. The recommended dose rates for common species in
South Africa are listed in Table 4. Dosage depends on the degree of effect required. Nervous and highly
excitable animals require a higher dose, whereas in older animals, or those that have undergone severe
physical exertion, the effect of xylazine is more pronounced. As the effect of xylazine can be influenced
by noise, disturbance, handling, etc., the animal should be left undisturbed until the effect is fully
established. If the effect is not adequate, at least 30-60 minutes must elapse before a further injection is
made.
Side-effects and special precautions. Xylazine has a variable effect on the cardiovascular system. In many
species an initial period of hypertension is followed by a longer period of hypotension. Bradycardia and
second-degree atrioventricular (AV) blocks are commonly encountered. Xylazine appears to sensitize
the heart to adrenaline and must, therefore, be used with caution when used together with halothane.
Drooping of the head is characteristic in the horse: this may result in hypostatic congestion of the nasal
mucosa.
Salivation, rumeno-reticular atony, and bloat occurs commonly in all ruminants. These animals should
not be left in lateral recumbency following immobilization. A liquid, sometimes haemorrhagic,
diarrhoea may occur up to 18-24 hours after use. Vomition is commonly observed in cats, and
occasionally in dogs.
A pronounced, persistent hyperglycaemia, due to increased hepatic glucose production and decreased
plasma insulin activity, occurs. Polyuria and glucosuria lasting for up to six hours frequently follows.
Thermoregulation is affected, and hyper- or hypothermia may result, which may last for up to 24 h.
Animals must be protected against chilling or exposure to high ambient temperatures.
Xylazine should be used with care in debilitated animals, in animals with depressed respiration or
pre-existing cardiac conduction disturbances, in cases of depressed hepatic and renal function, in shock,
and in animals with urethral obstruction.
When xylazine is used as the sole immobilizing agent animals may be aroused, potentially jeopardizing
the safety of the operator. The analgesic effect is variable, and its extent should be ascertained before
any clinical procedure is begun: a mildly sedated animal may make effective use of defence mechanisms
if pain is inflicted. Used in combination with anaesthetic and opioid drugs, the effects of xylazine are
additive. In such instances, dosages should be reduced and caution exercised.
Reversal of drug effects. The sedative effects of xylazine can be reversed with specific
alpha-2-adrenoceptor antagonists. Yohimbine (Yohimbine®, Milborrow Animal Health) is
recommended at 0,125 mg/kg. Xylazine-induced immobilization is also rapidly reversed by 1 mg of
atipamezole (Antisedan®, Ciba Geigy) for every 8-12 mg of xylazine used38. The analeptic drugs
doxapram (Dopram®, Continental Ethicals) at 1-2 mg/kg, and 4-aminopyridine (Kyron) at 0,3-0,6
mg/kg, have also been used successfully to hasten recovery. 4-Aminopyridine should not be mixed with
yohimbine, but should be injected separately: the two substances are chemically incompatible and
precipitate, forming a cloudy solution, when mixed in the same syringe. See details on antidotes in
Section 8 below.
Toxicity and particulars of its treatment. Respiratory depression and second- to third-degree AV-blocks
are the most important toxic effects that occur with overdosage of xylazine. These effects can be
reversed with the use of the specific antidotes and analeptic drugs. Atropine at 0,04 mg/kg is used to
treat the AV-block.
Stability and storage instructions. Xylazine is very stable. It should not be stored at above 30°C.
4.2.2 Detomidine
Detomidine (Domosedan®) (4-(2,3-dimethylphenyl)methyl-1H-imidazole hydrochloride) is available as
a 10 mg/ml injectable solution. It is manufactured by Farmos and supplied by Ciba Geigy in South
Africa.
Chemical and physical characteristics. Detomidine is an almost white, crystalline powder that is readily
soluble in water, ethanol, methanol, and chloroform. The aqueous solution is clear and colourless, with a
pH of 4-5.
Pharmacological action. The pharmacological effects of detomidine are typical of those described for the
group. However, detomidine is considerably more potent as an agonist of the alpha-2-adrenoceptors
than xylazine82. Only at high doses does the action on alpha-1-adrenoceptors occur. Xylazine appears to
produce more of an alpha-1-adrenoceptor effect: this may explain the pharmacological differences
between detomidine and xylazine. High doses of detomidine produce deep sedation, leading to loss of
consciousness, and a light plane of anaesthesia36. In laboratory studies, higher doses have been found to
lead to a decrease in sedation and an increase in sedative-analgesic activity, while hypnosis does not
occur. Detomidine does not induce a loss of the righting reflex of animals at any dose level84.
Detomidine is rapidly and completely absorbed after subcutaneous or intramuscular injection. It is
highly lipid soluble, resulting in very rapid distribution to the brain. After extensive distribution the
drug is metabolized in the liver and excreted, the majority of the metabolites passing into the urine with
a small fraction excreted in the faeces85. The elimination half-life is one to two hours. The duration of
action of detomidine in the horse is longer than that of xylazine, being up to six hours.
Indications. Detomidine is indicated as a potent sedative and analgesic (somatic and visceral) in the
horse, the sedative and analgesic effects being dose dependent in depth and duration. It can be used on
its own in horses to facilitate handling, transportation, examination, and minor surgical procedures, or
as a premedication to both injectable and inhalant anaesthesia. It has also been used in a 'cocktail' with
various opioid drugs. In dogs detomidine has unpredictable (not dose-related) sedative effects82.
Detomidine has been used successfully to sedate various species of wild animals, such as gemsbok, black
and blue wildebeest, eland, red hartebeest, zebra, and giraffe, and for immobilization in combination
with etorphine86.
Dosage and directions for use. Detomidine can be administered by intramuscular or slow intravenous
injection. The recommended dose rate is 20 µg/kg, but may vary from 20-150 µg/kg depending on the
depth and duration of sedation required. The dose rates for the anticipated level of sedation, dose
volume, commencement of effect, and duration of action are summarized in Table 5. Individual animals
appear to be tolerant to the effects of detomidine: this may, however, be due to underdosing. If needed,
1-15 times the initial dose may be repeated with no untoward effects. The desired effects usually develop
in 2-10 minutes.
Detomidine has been combined with opioids, such as morphine, pethidine, and butorphanol, to increase
its sedative and analgesic effects.
Side-effects and special precautions. The side-effects of detomidine are similar to those described for
xylazine. Rise in blood pressure is greater and longer lasting with higher doses. The incidence of
arrhythmias appears to be lower after intramuscular injection than after intravenous administration.
Other side-effects include sweating, snoring, and transient tremors.
Reversal of drug effects. Any of the alpha-2-adrenoceptor antagonists can be used to reverse the effects of
detomidine. Atipamezole (Antisedan®, Ciba Geigy) can be used at 4-6 times the preceding detomidine
dose.
Toxicity and particulars of its treatment. Respiratory and cardiac depression are associated with
overdosage. These effects are normally transient and reversible. Specific antagonists and analeptic
drugs could be used for treatment.
Stability and storage instructions. Store in a cool place below 25°C and protect from light.
4.2.3 Medetomidine
Medetomidine (Domitor®, Ciba Geigy) ((±)4-[1-(2,3-dimethylphenyl) ethyl]-1H-imidazole
hydrochloride) is available as a 1 mg/ml injectable solution in 10 ml multidose vials. It is currently in the
process of registration by Ciba Geigy in South Africa.
The chemical structure of medetomidine differs from that of detomidine by one methyl group in the
CH2-bridge. Its pharmacodynamic properties resemble those of detomidine, although it is more potent
and its mechanism of action is more selective.
Medetomidine is a racemic mixture, the active isomer being predominantly the d-enantiomer
(dexmedetomidine)1. Characteristic effects of medetomidine in different laboratory animals include
sedation, analgesia, relief of anxiety, bradycardia, hypotension, and hypothermia38. At high doses it has
hypnotic or anaesthetic effects.
The pharmacokinetics of medetomidine, as determined in rats, cats and dogs, indicate rapid absorption
and distribution of this highly lipophilic drug87. Peak concentrations are reached 30 minutes after
intramuscular injection. Medetomidine is metabolized in the liver, and the metabolites are excreted in
the urine. In rats, elimination is controlled by the extent of enterohepatic circulation. Significant
presystemic elimination occurs following oral administration.
Medetomidine has been tested extensively in a large number of different non-domestic mammals38,88.
In combination with ketamine it has been used for immobilization, and alone it induces sedation in a
dose-dependent manner. Complete immobilization has been achieved in semi-domesticated reindeer
and blue foxes38. In combination with ketamine, medetomidine doses are usually 60-100 µg/kg.
Remarkably low doses of ketamine are given in these combinations.
The alpha-2-adrenoceptor antagonist, atipamezole, is highly effective in reversing the immobilization
induced by medetomidine and medetomidine-ketamine combinations. In ruminants, the atipamezole
dose is 4-5 times the medetomidine dose used. An initial dose of 100-150 µg/kg is given intravenously and
the remainder is given subcutaneously. Because the required ketamine doses are relatively high in
carnivores, only half the ruminant dose of atipamezole is suggested in these species when medetomidine
is used in combination with ketamine. Using this dose, reversals are calm, and animals show minimal
'residual ketamine effect'. The dose of atipamezole in dogs is 4-6 times and in cats 3-4 times the
preceding dose of medetomidine, when medetomidine is given alone. Medetomidine effects are reversed
in 5-10 minutes. Smaller doses of atipamezole are required the longer the interval becomes between
medetomidine injection and the administration of the antagonist.
Increased future use of medetomidine and atipamezole in the immobilization, capture, and sedation of
wild animals is anticipated.
5 Tranquillizers/neuroleptics
Neuroleptics or tranquillizers are psychotropic agents that result in the suppression of behavioural responses without
affecting spinal and other reflexes89. Many other terms have been used, one of the most widely used being ataractic
(meaning perfect peace, calmness of mind, not disturbed). A characteristic of these drugs is that neuroleptic effects are
generally observed at low doses, and that higher doses induce characteristic cataleptic immobility. Arousal from CNS
depression effects is easily accomplished, and even at very high doses most tranquillizers do not induce coma89. A large
number of neuroleptic drugs are available, of which only three groups, viz. phenothiazines, butyrophenones, and
thioxanthenes, are used in wild animals. Thioxanthenes include zuclopenthixol and flupenthixol: these are discussed in the
next section on long-acting tranquillizers.
Neuroleptic drugs are all listed as Schedule 5 substances (Act 101, 1965).
5.1 Phenothiazine derivatives
Phenothiazine derivatives are multipotent receptor blockers that give rise to a large number of pharmacological
responses35. The receptor-mediated responses can be ascribed to either their effects on central
neurotransmission, or to their influences on neuronal terminal junctions in peripheral tissues35,90,91,92.
Centrally, phenothiazines are postsynaptic antagonists, and they inhibit catecholamine re-uptake at neuronal
terminals. The antagonism in the basal ganglia results in neurolepsis, while antipsychotic effects arise from action
in the limbic system and hormonal side-effects from activity in the hypothalmus. Peripheral responses to these
drugs are as a result of predominantly an adrenergic blockade. They are often complex in their pharmacological
interpretation, as neuronal re-uptake, which reduces catecholamine-mediated responses, is inhibited at the same
time. Peripheral and central cholinergic, antihistaminic, and antiserotonin effects also contribute to the
peripheral manifestations89. Some of the specific responses seen after injection of phenothiazine derivatives are
summarized in Table 6. Acepromazine and propionylpromazine are the predominant drugs of this group used in
wild animals, although chlorpromazine and promazine have been used in the past. Perphenazine enanthate and
pipothiazine palmitate are long-acting phenothiazine neuroleptic drugs that recently have been used more
extensively in wild animals (see the next section).
5.1.1 Acepromazine
Acepromazine maleate (Aceprom®, Milborrow Animal Health; Combistress®, Phenix; Neurotranq®,
Krüger-Med) (2-acetyl-10-(3-dimethylaminopropyl)phenothiazine) was formerly known as
acetylpromazine. It is available for veterinary use as 2, 10, and 20 mg/ml injectable solutions and as 10
and 25 mg tablets. Aceprom® 2 mg and 10 mg/ml injectable solutions are supplied in 20 ml clear rubber
capped vials. Combistress® is supplied as a 20 mg/ml injectable solution and Neurotranq® as a 10
mg/ml injectable solution in 50 ml multidose amber glass vials. Aceprom® tablets are packed in
quantities of 100.
Chemical and physical characteristics. Acepromazine is an acetyl derivative of promazine. The powder is
yellow, odourless, and crystalline with a bitter taste. Thirteen-and-a-half milligrams of acepromazine
maleate is approximately equivalent to 10 mg of acepromazine. The powder is soluble at a ratio of 1 to
27 in water: stronger solutions of up to 40 mg/ml tend to crystallize on standing and cooling, but have
nevertheless been used23. Solutions of 100 mg/ml have been made up in propylene glycol.
Pharmacological action. Most of the pharmacological effects of acepromazine are similar to those
described for the group. It is more potent than the other derivatives used in wild animals, and is effective
parenterally in small doses.
Indications. Acepromazine has been used extensively for tranquillization, neuroleptanalgesia (in
combination with opioids particularly etorphine), and as a premedication for general anaesthesia in
wild animals. It is seldom used for immobilization on its own. In combination with opioids,
cyclohexylamines, or other anaesthetic agents, it has synergistic activity, lessening undesirable
side-effects, shortening induction time, and reducing the dose of the particular immobilizing drug with
which it is combined.
Dosage and directions for use. Acepromazine is recommended at 0,125-0,25 mg/kg in dogs and cats and
at 0,05-0,1 mg/kg in horses, cattle, and sheep. Injectable solutions are administered intramuscularly or
by slow intravenous injection. The dose rate of tablets is 1-3 mg/kg, orally.
Acepromazine is seldom used alone but rather in combination with either opioids or anaesthetic agents.
Since synergistic action is achieved with these immobilizing drugs, combinations must be used with
caution: it may be necessary to reduce the dose of the immobilizing drug by approximately one half.
A slight drooping of the upper eyelid, associated with partial protrusion of the nictitating membrane,
are the earliest signs to occur following administration of acepromazine. The onset of action after
intravenous injection is 10-20 minutes. For optimum effect, animals should not be subjected to sensory
stimuli whilst the drug is taking effect. The duration of action varies between species: in general, action
is prolonged, lasting for 4-8 hours. Residual sedation may last for 12 hours.
It is contra-indicated to mix glycopyrrolate in the same syringe with acepromazine35.
Side-effects and special precautions. The side-effects of acepromazine are typical of the phenothiazine
group: these are listed in Table 6. Due to the hypotensive effects of the drug it must be used with caution
in weak, debilitated, or aged animals, and in animals with cardiac disease. In most types of shock the use
of acepromazine could result in cardiovascular collapse due to its alpha-adrenergic blocking action: if
possible, fluid therapy should commence before the administration of acepromazine in these patients.
The disruption of the thermoregulatory mechanism caused by acepromazine could result in
hyperthermia or hypothermia, depending on the ambient temperature. The body temperature must,
therefore, be monitored regularly, and animals should be protected against heat and cold.
Hyperthermia has resulted in fatalities, especially in shade-loving animals such as the waterbuck23.
Acepromazine should not be administered to animals when the ambient temperature is high.
Tranquillization of dangerous animals may lead to a false sense of security. Painful procedures should
be avoided if acepromazine is given alone, since phenothiazines provide little, if any, analgesic effect.
Occasionally, the use of acepromazine results in a paradoxical hyperexcitement and hyperaesthesia,
especially in the horse. It may also induce aggression in dogs.
Extrapyramidal effects, leading to muscle tremors that may progress to stiffness and muscle spasms, are
also observed occasionally.
Reversal of drug effects. There is no specific antidote to acepromazine. Doxapram (Dopram®,
Continental Ethicals) has been found to reverse effects of phenothiazine derivatives effectively.
Toxicity and particulars of its treatment. Mortality is as a result of cardiovascular and respiratory
collapse. Treatment of overdosage is symptomatic and supportive. Doxapram® (Dopram, Continental
Ethicals) has been effective in reversing the effects of acepromazine in the dog. Dopamine
(Dopamin-Natterman®, Natterman; Intropin®, Boots; Sabax Dopamin®, Sabax) and dobutamine
(Dobutrex®, Eli Lilly) can be used to stimulate cardiac function. These very potent drugs must be
diluted in non-alkaline solutions before administration. Adrenaline is contra-indicated following
acepromazine administration due to additional vasodilatory effects by beta-2-adrenoceptor action.
Some people may suffer dermal photosensitization reactions if the solution is handled carelessly.
Stability and storage instructions. Store in a cool, dry place below 25°C.
5.1.2 Propionylpromazine
Propionylpromazine hydrochloride (Combelen®, Bayer Animal Health)
(2-propionyl-10-(3-dimethylaminopropyl)phenothiazine) is available as an injectable solution of 10
mg/ml. It is supplied in cartons containing 5 x 25 ml bottles.
Chemical and physical characteristics. Propionylpromazine hydrochloride is an odourless, yellow
powder that is readily soluble in water. The injectable solution is clear and yellow, and contains
methyl-p-hydroxybenzoate 0,10% m/v and sodium sulphite 0,03% m/v as preservatives.
Pharmacological action. Similar to acepromazine. It is extensively metabolized in the liver to at least
three metabolites.
Indications. Propionylpromazine is used for the immobilization of various wild animal species in
combination with particularly opioids but also with the cyclohexylamines. It is also used as a
tranquillizer following physical capture and for transportation of certain antelope species.
Dosage and directions for use. Propionylpromazine is recommended at a dose rate of 0,03-0,2 mg/kg,
administered by slow intravenous injection or intramuscularly. Like acepromazine, it potentiates the
pharmacological effects of opioids and cyclohexylamines when used in combination with either of these
drugs: lower doses of these drugs are consequently required.
Side-effects and special precautions. Similar to acepromazine. However, propionylpromazine is reported
to have a greater tendency to cause an irreversible paralysis of the penis in stallions, particularly
following castration35. Localized swelling at the site of injection may occur following subcutaneous
injection.
Reversal of drug effects. There is no specific antidote to propionylpromazine. Doxapram (Dopram®,
Continental Ethicals) has been found to reverse effects of phenothiazine derivatives effectively.
Toxicity and particulars of its treatment. Excessive sedation and a subsequent drop in blood pressure are
the major effects seen following overdosage. Treatment is symptomatic and supportive. Dopamine
(Dopamin-Natterman®, Natterman; Intropin®, Boots; Sabax Dopamin®, Sabax) and dobutamine
(Dobutrex®, Eli Lilly) are suggested for treatment of cardiac failure. These drugs are very potent
cardiac stimulants and must be diluted in non-alkaline solutions before administration. Doxapram
(Dopram®, Continental Ethicals) is recommended for stimulation of respiration.
Stability and storage instructions. Do not store above 25°C.
5.1.3 Chlorpromazine and promazine
Chlorpromazine (Largactil®, Maybaker) and promazine (Sparine®, Akromed) were used for the
tranquillization and capture of exotic species in the past. They have largely been replaced by the more
potent acepromazine and propionylpromazine drugs of this group.
5.2 Butyrophenones
Butyrophenones produce their effects through a central dopaminergic and a peripheral adrenergic blockade. In
the extrapyramidal system, butyrophenones may act by mimicking action of gamma-aminobutyric acid (GABA),
or by preventing the effect of glutamic acid on synaptic junctions35. Peripheral adrenergic effects generally only
occur at higher doses35. The specific responses seen after butyrophenone-derivative administration are
summarized in Table 7. Butyrophenones used in wild animals are azaperone, droperidol, and haloperidol.
Droperidol is used in combination with fentanyl as a neuroleptanalgesic, and has been discussed in the section on
the opioids. Haloperidol is most commonly used to provide long-term tranquillization: it is therefore discussed in
the section on long-acting tranquillizers.
5.2.1 Azaperone
Azaperone (4'-fluoro-4-[4-(2-pyridyl)-1-piperazinyl]butyrophenone) is available as Stresnil® from
Janssen Pharmaceutica in an injectable solution of 40 mg/ml. Azaperone powder is not readily available,
but can be obtained directly from Janssen Pharmaceutica on request.
Chemical and physical characteristics. Azaperone is a yellowish to slightly tan, amorphous or crystalline
powder that dissolves easily in an acid medium. Azaperone base and solutions are affected by light.
Solutions have a pH of 4,4 or higher, depending on the dilution.
Pharmacological action. The pharmacological effects of azaperone are similar to those described for the
butyrophenones. Following intravenous administration an increase in respiratory rate has been
observed in dogs, pigs, rats, and black bears24. Azaperone has been found to counteract the respiratory
depression caused by fentanyl.
Azaperone is a short-acting drug. It is active within 15-30 minutes, lasts for 2-3 hours, and is nearly
eliminated from body tissues within 16 hours. The primary site of metabolism is the liver.
Indications. Azaperone is recommended for the sedation and immobilization of a wide variety of wild
animals. It is used in combination with either opioids or anaesthetic agents for immobilization.
Dosage and directions for use. When used on its own azaperone is effective at doses from 0,5-2,0 mg/kg
body mass. When combined with etorphine or fentanyl, the dosage can be reduced to as low as 0,05
mg/kg. As a general guide, azaperone is used at 0,5 mg/kg body mass in herbivores and 1,0 mg/kg in
carnivores93. Lower doses are used in the wild where the animal needs to regain normal flight responses
rapidly because of the presence of predators. For use in immobilizing mixtures with opioids, an average
dose level of 0,15-0,3 mg/kg is used for most herbivorous species, except zebra, warthog, impala,
steenbok, and springbok, where 0,3-0,5 mg/kg is advocated93.
A solution of 100 mg/ml azaperone is used mostly for immobilization mixtures. This is prepared by
combining 0,5 g tartaric acid with 10 g azaperone powder and 20 ml distilled water. This is warmed to
80°C in a warm bath, made up to 100 ml, and filtered. The solution can be concentrated to 200 mg/ml,
but this is thick and 'syrupy', and is difficult to work with93. Stresnil® (Janssen), which contains 40
mg/ml azaperone, is also used effectively in wild animals.
Side-effects and special precautions. Similar to those described for the phenothiazine group. The severity
of the drop in blood pressure is related to dose level, and occurs within 5-10 minutes after
administration. At low dosages, azaperone does not appear to have an adverse effect on
thermoregulation, heart rate, cardiac output, or aortic blood flow. It has a moderate beta-adrenoceptor
action, and may suppress sympathetic reflexes.
Extra-pyramidal signs and allotriophagia are sometimes observed following the use of azaperone.
Reversal of drug effects. There is no known antidote to azaperone.
Toxicity and particulars of its treatment. The safety margin between the lowest and the lethal dose is
stated to be a factor of more than 100023. Treatment of overdosage is symptomatic and supportive.
Stability and storage instructions. Azaperone base and solutions remain stable for three to four years
when stored in the dark. Similar stability is claimed in combination with fentanyl. Temperatures of up
to 40°C do not affect stability.
6 Long-acting tranquillizers
The use of depot, or long-acting, tranquillizers has been described as a new concept and an important breakthrough in the
management and translocation of wildlife94. These drugs are used to reduce anxiety and stress during long-distance
translocation by road or air, for acclimatizing or adapting wild animals recently captured and introduced into foreign and
unnatural situations, enclosures and new habitats, and to sedate animals sufficiently to enable them to withstand the
stressful activities at game auctions.
Long-acting tranquillizers have been defined as tranquillizers that can be injected in such a form and manner that a single
dose gives effective tissue concentration for at least a week95. While haloperidol does not conform to this definition, it is
included in this section because haloperidol is used in wild animals when sedation for up to 16 hours is required:
haloperidol is effective in animals for this length of time, i.e., more than twice the length of effectivity of other
tranquillizers. Long-acting tranquillizers available in South Africa are listed in Table 8.
Prolonged action of tranquillizers has been achieved by either sustained release (depot) formulations, or by the use of
products with slow metabolism and elimination of the active ingredient. Most long-acting tranquillizers have been
esterified and are formulated in a vegetable oil: these induce delayed metabolism and delayed absorption, respectively. As a
result of delayed absorption it may take several hours to a few days before the onset of long-acting neuroleptic effect. It is
therefore necessary to 'load' or initiate the process by injecting a short-acting tranquillizer concurrently with the
long-acting tranquillizer. Haloperidol has proved to be an ideal drug for this purpose when used in conjunction with
long-acting tranquillizers94. A combination of long-acting tranquillizers such as perphenazine enanthate (Trilafon-LA®)
and zuclopenthixol acetate (Clopixol-Acuphase®) compliment the pharmacokinetic effect of each drug in terms of onset
and duration of effect. For example, the calming effect of the zuclopenthixol acetate starts within an hour of injection and
by the third day, when its effect starts wearing off, the effect of the perphenazine reaches its peak94. A summary of the
onset and duration of effect of haloperidol and various long-acting tranquillizers used in wild animals is given in Table 9.
Animals under the influence of long-acting tranquillizers must be handled quietly and calmly, and with caution:
unnecessary noise and activity may arouse these animals and result in injury to both animal and handler.
Extrapyramidal symptoms are the most important side-effects of long-acting tranquillizers in humans. These include
akathisia (i.e., hyperactivity, incessant restlessness, and anxiety), akinesia (i.e., weakness, muscular fatigue, and reduction
of voluntary activity), dyskinesia (i.e., dystonic reactions such as torticollis and opisthotonus, spasmodic bodily movements,
facial grimacing, and involuntary rhythmic movements), parkinsonism (i.e., rigidity of limbs, tremors, facial rigidity, and
posture disturbances), and persistent tardive dyskinesia (i.e., repetitive, uncontrolled movements of the tongue and
mouth)94.
Long-acting tranquillizers should be used with caution when combined with drugs that cause central nervous system
depression. They may also increase the occurrence of extrapyramidal effects if used in conjunction with other
anti-dopaminergic agents. It must be remembered that all phenothiazines interfere with thermoregulation: animals being
transported while under the influence of long-acting tranquillizers are therefore likely to be susceptible to chilling or
overheating. Phenothiazines can also lower the seizure threshold in susceptible individuals.
All long-acting neuroleptic products and haloperidol are listed as Schedule 5 substances and require a veterinary
prescription for use in animals. None of these products are currently registered for veterinary use.
6.1 Tranquillizers with a long action
6.1.1 Haloperidol
Haloperidol (Serenace®, G. D. Searle)
(4'-fluoro-4-[4-hydroxy-4(4-chlorophenyl-0-piperidino]-buterophenone) belongs to the butyrophenone
group of neuroleptics with a longer duration of activity. It is available as 5 and 10 mg/ml injectable
solutions in 1 and 2 ml ampoules, respectively. The ampoules are supplied in packs of six. Haloperidol as
a decanoate ester in an oily depot formulation was previously available as 50 and 100 mg/ml injectable
solutions, but has subsequently been withdrawn from the market.
Chemical and physical characteristics. Haloperidol is a white to faintly yellowish, odourless, amorphous
or microcrystalline powder. It is practically insoluble in water. In the presence of 1% lactic acid or
tartaric acid, stable solutions in water can be prepared containing up to 20 mg/ml. The solutions have a
pH of about 3,2 and are slightly hypo-osmotic.
Haloperidol is incompatible with etorphine and fentanyl, and can therefore not be incorporated in
neuroleptanalgesic mixtures. Solutions of haloperidol can also not be mixed with heparin in glucose or
sodium chloride injections.
Pharmacological action. The pharmacological effects are similar to those described for the
butyrophenone tranquillizers, but it is generally more potent. In contrast to the less potent sedative
neuroleptic drugs, such as promazine, the actions of haloperidol are more specific. The antipsychotic
effect has been described as being associated with the inhibitory effects on the dopaminergic
nigrostriatum system of the midbrain, and the phychomotor sedative effects as being associated with the
inhibitory effects on the noradrenergic median forebrain bundle system96. Fewer peripheral effects,
such as hypotension and poikilothermia, are observed. There is some evidence that haloperidol possesses
analgesic properties, but at dosages affecting spontaneous activity and motor coordination the analgesic
effect may be unspecific96.
Onset of effect after intravenous injection is usually within 5-10 minutes. Depending on the dose and
species, the effective duration varies from 8-18 hours.
Indications. Haloperidol is recommended for sedating highly excitable, nervous, and aggressive species
for translocation procedures. These include calming immediately after capture, transportation,
temporary holding in pens or bomas, quarantine, auctions, loading and off-loading, relocation, and
adaptation to new surroundings97. In these situations, haloperidol may be used on its own or for initial
sedation in a long-acting tranquillizer regime. The product has been shown to be particularly effective
in the majority of small- and medium-sized antelope species, especially red hartebeest, blesbok,
springbok, duiker, steenbok and dik-dik, and possibly tsessebe96. In larger ungulates, variable results
have been obtained at high (0,3 mg/kg) doses: favourable responses have, however, been recorded in
young kudu, sable calves, Burchell's zebra, and Hartmann's zebra96. Haloperidol has also been
reported to be useful and effective for the transportation of black rhinos97.
Dosage and directions for use. Haloperidol can be injected intravenously or intramuscularly. The dosage
rate varies considerably between species: consequently, in many cases animals are overdosed.
Recommended doses for free-ranging animals are: Burchell's zebra (0,3 mg/kg), Hartmann's zebra
(0,28-0,35 mg/kg), blesbok (0,11-0,28 mg/kg), tsessebe (0,25-0,3 mg/kg), black-faced impala (0,08-0,10
mg/kg), springbok (0,2-0,3 mg/kg), duiker (0,45 mg/kg) and steenbok (0,48 mg/kg)96,99. In red
hartebeest the following dosage rates have been used: boma-captured (0,13-0,16 mg/kg), netted (0,08-0,1
mg/kg), and captive (0,06-0,08 mg/kg)96. Animals must be kept calm and quiet following administration
of haloperidol.
Side-effects and special precautions. Extrapyramidal symptoms can potentially occur in any animal, but
are seen more frequently in roan antelope, black-faced impala, red hartebeest and, to a lesser degree, in
blesbok and duiker96. Springbok tend to show a transient restlessness. Hypertonia and allotriophagia
are more commonly observed in captive than in free-ranging animals. An increased tendency to develop
enhanced extrapyramidal symptoms appears to occur with overdosage, hyperthermia, noise,
excitability, and a concomitant catecholamine reaction. Care should, therefore, be taken not to
over-excite animals during capture and handling, and not to exceed recommended dosage rates in
species that are prone to these symptoms. In view of the abnormal feeding behaviour that occasionally
accompanies haloperidol therapy, special care should be taken to prevent the ingestion of foreign
material.
Mild soporific effects and ataxia may occur in some species such as in Hartmann's zebra and
black-faced impala. Haloperidol is almost devoid of peripheral autonomic effects at normal doses.
Large- and medium-sized herbivores tranquillized with haloperidol should be handled with caution as
they tend to show aggression towards people and may become extremely dangerous when on their own.
When kept individually in pens or crates, adult kudu bulls, adult male hartebeest, and blesbok rams
may attack without provocation when under the influence of haloperidol96.
The effects of haloperidol decanoate were generally unsatisfactory since it resulted in inappetance and
prolonged sedation.
Toxicity and particulars of its treatment. Overdosage of haloperidol can result in severe hypotension.
Treatment is symptomatic and supportive. Extrapyramidal symptoms can be treated with biperidine
(Akineton®, Knoll) or diazepam (Valium®, Roche).
Stability and storage instructions. Haloperidol solutions are stable except when exposed to light: exposed
solutions become discoloured after a few hours, and deposit a greyish-red precipitate after several
weeks.
6.2 True long-acting tranquillizers
6.2.1 Zuclopenthixol
Zuclopenthixol acetate (Clopixol-Acuphase®, Lundbeck) ((Z)-2-{4[3-(2-chloro-10H-dibenzo[b,e]thiin-10-ylidene)propyl]pipirazin-1-yl}acetate) is available as a 50 mg/ml
sterile vegetable oil (viscoleo) solution. It is supplied in packs of 5 x 2 ml ampoules. Zuclopenthixol
decanoate (Clopixol Depot®), a 200 mg/ml oily solution, is also available.
Chemical and physical characteristics. Zuclopenthixol is a cis and trans racemic mixture. The substance
has been esterified with either acetate or decanoate to achieve longer action.
Pharmacological action. Zuclopenthixol is a thioxanthene with general properties similar to the
phenothiazine tranquillizers. Its duration of effect has been extended by esterification, which delays
metabolism and release of the active component, and by dissolving in a vegetable oil to delay absorption.
The onset of effect of zuclopenthixol acetate given intramuscularly occurs within one hour, and the
effect lasts up to 3-4 days94. In the case of the decanoate, the duration is extended to 10-21 days. The
sedative effects of zuclopenthixol decanoate were found inadequate for use in wild animals (H. Ebedes,
pers. comm.).
Indications. Zuclopenthixol acetate is used as an intermediate long-acting tranquillizer for the sedation
and calming of a variety of herbivores. It is used either alone or to bridge the effects of shorter and
longer acting neuroleptic drugs when used in combination94.
Dosage and directions for use. Zuclopenthixol acetate must be given either by deep intramuscular
injection or subcutaneously. A total dose of 50-100 mg has been used in a number of different
free-ranging and captive wild animals. It can be used alone or concurrently with longer acting drugs
such as perphenazine enanthate (Trilafon LA®) or pipothiazine palmitate (Piportil Depot®,
Maybaker). Animals must be handled calmly and quietly during the onset of effects.
Side-effects and special precautions. Very few side-effects are observed. In some animals, in the early
phase of treatment and at higher doses, extrapyramidal symptoms may occur. Some animals may show
autonomic side-effects. Overdosage may also result in inappetance.
Toxicity and particulars of its treatment. Overdosage may lead to somnolence, coma, convulsions, and
circulatory collapse. Extrapyramidal effects can be treated with antiparkinsonian drugs such as
biperidin (Akineton®, Knoll). Convulsions and muscular spasms can be treated with diazepam
(Valium®, Roche).
Stability and storage instructions. Store below 25°C and protect from light.
6.2.2 Perphenazine
Perphenazine enanthate (Trilafon®, Sherag)
(2-{4-[3-(2-Chlorophenothiazin-10-yl)propyl]piperazin-1-yl}ethanol) is available as a 100 mg/ml
solution in sesame oil. It is supplied in 1 ml ampoules.
Chemical and physical characteristics. Perphenazine powder is white or creamy-white and is odourless or
almost odourless. It is practically insoluble in water. Perphenazine enanthate is a long-acting ester
contained in a sesame oil vehicle. Each ml of 100 mg/ml perphenazine enanthate is equivalent to 78,3 mg
perphenazine. The injectable solution has a pH of 4,5-5,6.
Pharmacological action. Perphenazine is a phenothiazine derivative with a piperazine side chain. It has
properties similar to those described for the phenothiazine tranquillizers. It has actions at all levels of
the central nervous system, particularly the hypothalmus, and demonstrates anxiolytic, anti-psychotic,
and anti-emetic properties. In animals, operant behaviour is reduced, spontaneous motor activity is
diminished, and conditioned avoidance behaviours are selectively inhibited, while unconditioned escape
or avoidance responses are not altered.
The prolonged action of perphenazine enanthate is attributed to the formation of a tissue oil depot, from
which the drug is slowly released, and to hydrolysis of the ester to form the free base. Perphenazine is
extensively biotransformed in the liver, principally by sulphoxide formation, hydroxylation of the
tricyclic ring, and dealkylation of the piperazine ring. Most of the drug and metabolites are excreted in
the faeces.
The onset of effect of perphenazine enanthate is slow, with sedation and calming effect in wild animals
first noted from about 12-16 hours after injection94. Maximum or peak effect is usually observed on the
third day, with duration of effect being up to seven days.
Indications. Indicated as a long-acting tranquillizer for the sedation and calming of a variety of
herbivores94.
Dosage and directions for use. Perphenazine enanthate is administered by deep intramuscular injection.
Intravenous injection of the oily enanthate solution is contra-indicated. A total dose of 20-200 mg is
administered depending on the size of the animal. Since extrapyramidal symptoms increase in
frequency and intensity with increased dosage, the lowest effective dose should be administered.
Zuclopenthixol acetate may be given concurrently with perphenazine enanthate to achieve sedation
during the initial 2-3 days, before perphenazine takes full effect.
Side-effects and special precautions. Similar to zuclopenthixol acetate. Adverse effects with overdosage
are similar to those described for the phenothiazine tranquillizers.
Concurrent administration of perphenazine enanthate may potentiate central nervous system
depressant effects of opioids, barbiturates, or other sedatives, anaesthetics and neuroleptics, and the
anti-cholinergic effects of atropine and antihistamines. Respiratory depressant effects of pethidine and
other opioids may also be increased.
Toxicity and particulars of its treatment. As for the phenothiazine tranquillizers. Treatment of
extrapyramidal symptoms and convulsions is the same as for zuclopenthixol.
Stability and storage instructions. Store at 2-30°C. Keep package closed to protect from light: exposure to
light may cause discolouration. If markedly discoloured, the ampoule should be discarded.
6.2.3 Pipothiazine
Pipothiazine palmitate (Piportil Depot®, Maybaker) (10-{3-4-(2-Hydroxyethyl)piperodino}
-NN-dimethyl-phenothiazine-2-sulphonamide palmitate) is available as a 50 mg/ml injectable solution in
sesame oil. It is supplied in 1 ml and 2 ml ampoules.
Pharmacological action. Pipothiazine palmitate is a long-acting phenothiazine tranquillizer with
pharmacological effects similar to those of perphenazine enanthate. Onset of sedation in wild animals is
delayed for up to 72 hours following administration of this product. Duration of effect is prolonged, and
may persist for 21-28 days.
Indications. Similar to perphenazine enanthate, but where longer effect is required.
Dosage and directions for use. Pipothiazine palmitate is administered by deep intramuscular injection.
Intravenous injection of the sesame oil solution is contra-indicated. A total dose of 20-200 mg is
administered depending on the size of the animal. Haloperidol or zuclopenthixol acetate is administered
with pipothiazine to achieve early sedation: with pipothiazine palmitate alone, sedation is only evident
from 48-72 hours after injection.
Side-effects and special precautions. Similar to perphenazine enanthate.
Toxicity and particulars of its treatment. Similar to perphenazine enanthate.
Stability and storage instructions. Store below 25°C and protect from light.
7 Neuromuscular blocking agents
Muscular activity can be affected by modifying the neurotransmission at the postsynaptic neuromuscular endplate.
Neuromuscular blocking agents cause relaxation and paralysis of muscles by interfering with the ability of acetylcholine
(ACh), the endogenous neurotransmitter, to activate nicotinic cholinergic receptors of the skeletal muscle cells. Distinct
subtypes of nicotinic receptors exist at the neuromuscular junction, and several pharmacological agents distinguish
between them100. Nicotinic cholinergic receptors of the autonomic ganglia may also be affected by neuromuscular
blocking agents.
Neuromuscular blocking agents were some of the earliest drugs used for immobilization of wild animals. They have
subsequently been replaced by more effective and safer drugs for use in most animals, but are still widely used for the
immobilization of reptiles. The current use of neuromuscular drugs for the culling of elephants remains controversial101.
When used as immobilizing drugs, it is important to consider that these agents do not anaesthetize animals, and are not
analgesic. Analgesia must be provided for any painful procedure performed on animals following restraint using these
drugs.
The safety margin between the effective and the toxic dose is very small with neuromuscular blockers. Respiratory failure,
as a result of paralysis of the respiratory muscles, occurs. Humans are particularly sensitive to these effects: extreme
caution must therefore be exercised with the use of these drugs. Although an antidote is available, respiratory support and
a supply of oxygen is essential in the case of accidental poisoning. These drugs are listed as Schedule 4 substances in the Act
and require a veterinary prescription for use in animals by non-veterinarians.
Neuromuscular blocking agents are distinguished by whether or not they cause depolarization of the motor end plate. For
this reason, they are classified either as competitive antagonists, such as gallamine, or as depolarizing agents, such as
suxamethonium. Both the examples mentioned are used in wild animals.
7.1 Competitive, non-depolarizing peripheral muscle relaxants
The members of this group are also referred to as the curare-type of agents, since the alkaloids of curare were the
first of these agents to be investigated. Competitive, non-depolarizing, muscle relaxants compete with ACh for
available nicotinic receptors at the postsynaptic membrane and, by occupying these receptors, prevent the
transmitter function of ACh. They then progressively reduce the ability of nerve action potentials to generate
endplate muscle action potentials at neuromuscular junctions. Some of these drugs have the ability also to reduce
the output of ACh by the nerve endings, possibly by acting on presynaptic cholinergic receptors.
The overall result is weakening and then paralysis of voluntary muscle, commencing in the face and then
progressing to the neck, limbs, abdomen, and finally the intercostal and diaphragmatic respiratory muscles.
Recovery takes place in the reverse sequence.
There are a number of drugs that are members of this group such as atracurium, d-tubacurarine, gallamine,
pancuronium, and vecuronium. Gallamine triethiodide (Flaxedil®) is currently the only drug used in the wildlife
field.
7.1.1 Gallamine
Gallamine triethiodide (Flaxedil®, Maybaker)
(2,2',2''-(Benzene-1,2,3-triyltrioxy)tris(tetraethylammonium) tri-iodide) is available as a 40 mg/ml
injectable solution. It is supplied in 2 ml and 3 ml ampoules.
Chemical and physical characteristics. Gallamine powder is white, or almost white, hygroscopic, and
odourless. The injectable solutions have a pH of 5,5 to 7,5. Solutions of gallamine triethiodide are
incompatible with thiopentone when the gallamine solution is added to thiopentone but not vice versa;
they are also incompatible with pethidine hydrochloride solutions68.
Pharmacological action. Gallamine is a competitive pharmacological antagonist of the post-junctional
neuromuscular nicotinic receptors. It also blocks the muscarinic receptors in the heart, and so improves
both blood pressure and heart rate. Onset of effect is within 1-2 minutes, and duration of action is 15-20
minutes. In crocodiles, the onset of action following intramuscular administration is 20-30 minutes, and
the duration of action is up to 12 hours. Immobilization and recovery are delayed in cold and inactive
crocodiles. Most of the drug is eliminated unchanged by the kidneys.
Indications. Gallamine triethiodide is indicated for the immobilization of reptiles, in particular
crocodiles102,103.
Dosage and directions for use. A dose of 0,4-2,0 mg/kg given intramuscularly in the base of the tail is
recommended for the immobilization of crocodiles. It produces a smooth induction without excitement.
Gallamine is not a general anaesthetic, and does not produce any analgesia. General or local anaesthesia
must therefore be given before any painful procedures are performed: without such anaesthesia severe
fright is seemingly evoked, and pronounced cardiovascular disturbances and even myocardial damage
may be caused.
Gallamine should not be administered together with any other agent having neuromuscular blocking
effects, such as aminoglycoside and polymixin antibiotics, clindamycin, lincomycin, and anaesthetic
agents such as halothane. Opioids produce additive respiratory depression. Other depolarizing and
non-depolarizing neuromuscular blockers should not be used simultaneously or sequentially before the
effect of the first is terminated.
Side-effects and special precautions. Hypertension and tachycardia occur at the onset of neuromuscular
blockade. Respiratory depression, apnoea, profound and prolonged muscle relaxation, and malignant
hyperthermia may occur as with any of the neuromuscular blocking agents. In crocodiles, a wide
therapeutic index (0,64-4,0 mg/kg) is observed, and only slight respiratory depression is evident. Other
side-effects of gallamine in crocodiles are mouth gaping and occasional copious salivation.
Reversal of drug effects. The effects of gallamine can be reversed with neostigmine methylsulphate
(Neostigmine®, Milborrow Animal Health; Prostigmin®, Roche) at 0,022 mg/kg.
Care should be taken that paralysis does not recur after antagonism by neostigmine; additional antidote
may be required. For unknown reasons, crocodiles sometimes do not react to neostigmine . Following
accidental human injection, atropine sulphate at 0,04 mg/kg is administered before the administration
of neostigmine to block the muscarinic side-effects of the antidote.
Toxicity and particulars of its treatment. Respiratory depression and apnoea caused by paralysis of the
respiratory muscles are the major effects of overdosage. Humans are particularly sensitive to the toxic
effects of this drug. Treatment is by administration of the antidote and atropine as mentioned under the
previous heading. In addition, mechanical ventilation and supply of oxygen are of the utmost
importance in humans. The use of respiratory stimulants such as doxapram is contra-indicated.
4-Aminopyridine, on the other hand, has been found to be effective as an antagonist of curare-induced
neuromuscular block35: it evokes release of acetylcholine from somatic nerve terminals and is longer
acting than neostigmine.
Stability and storage instructions. Store below 25°C and protect from light.
7.2 Non-competitive, depolarizing peripheral muscle relaxants
Drugs of this group interact with the pre- and post-synaptic nicotinic receptors at the neuromuscular junction.
Their effects are distinctly different to those of the curare-like drugs, initially eliciting an endplate potential and a
corresponding muscle action potential upon exposure. The depolarization changes in membrane potential are
similar to those produced by the endogenous mediator ACh. However, since depolarizing drugs are not
hydrolyzed as rapidly by cholinesterase as is ACh, they occupy the receptors for extended periods resulting in a
sustained depolarization. For as long as they remain attached to the nicotinic receptors, complete repolarization
of the postsynaptic membrane is prevented, thus rendering the motor end-plate nonresponsive to the normal
action of ACh. Subsequent impulse transmissions are blocked, and a flaccid type of paralysis ensues. Flaccid
muscular paralysis is preceded by involuntary and uncoordinated fibrillatory muscle contraction that, in
humans, may be moderately painful104.
Suxamethonium chloride (Scoline®), a member of this group, is used during game cropping operations and
occasionally for immobilization of reptiles.
7.2.1 Suxamethonium
Suxamethonium chloride (Scoline®, Glaxo) (2,2'-Succinyldioxybis-(ethyltrimethyl-ammonium)
dichloride dihydrate) previously known as succinyldicholine, is available as a 50 mg/ml injectable
solution. It is manufactured and supplied by Glaxo in South Africa. The product is not registered for
veterinary use.
Chemical and physical characteristics. Suxamethonium consists of two molecules of acetylcholine linked
at the alpha-methyl position. The powder is white or almost white, odourless or almost odourless,
hygroscopic, and crystalline. One milligram of suxamethonium is equivalent to 1,37 mg of
suxamethonium chloride dihydrate, and to 1,24 mg of anhydrous suxamethonium chloride. It is soluble
at a ratio of 1:1 in water and 1:350 in alcohol; it is practically insoluble in chloroform and ether. The
solution has a pH of 3,0-5,0. It is rapidly destroyed by alkalis and should, therefore, not be mixed with
alkaline injections such as thiopentone sodium.
Pharmacological action. The pharmacological effects of suxamethonium are similar to those described
for the group. Suxamethonium exerts very little effect on autonomic ganglia at normal doses. Its effects
following intravenous injections are seen within 15 seconds: these consist of irregular, uncoordinated
muscular contractions that last about half a minute, followed by flaccid paralysis of skeletal muscles.
Suxamethonium is normally rapidly hydrolyzed by liver and plasma pseudo-cholinesterase to an initial
metabolite, succinylmonocholine, which has a much weaker, predominantly competitive, type of
neuromuscular blocking action. This metabolite is then more slowly broken down by
pseudo-cholinesterase to succinic acid and choline. Potency of suxamethonium varies considerably
between species: this has been attributed to species differences in the activity of
pseudo-cholinesterase35. Cattle are, as an example, much more susceptible to the effects of
suxamethonium than horses: doses of about one-tenth of those used for the horse (0,125-0,20 mg/kg)
cause recumbency for about 15 minutes.
In crocodiles, the dose required to produce immobilization is usually very high compared with that for
mammals. However, dosage appears to be quite variable among species and sizes of crocodilians103. For
example, a dose of 3-5 mg/kg intramuscularly in juvenile American alligators produced muscle
relaxation in four minutes, with recovery complete in 7-9 hours. In Australian salt water crocodiles
under field conditions, doses of 5 mg/kg for 50 kg animals and 4 mg/kg for 200 kg animals resulted in
immobilization in 1 hour103. During induction, the coarse muscle twitches that occur are much stronger
than the fine tremors that occur in mammals. Once muscle function begins to return, recovery is rapid,
and the total immobilization time is generally less than the time crocodilians normally spend
submerged. Under the effects of suxamethonium, most animals continue to breathe, although chest
expansion is visibly decreased.
Indications. Suxamethonium is used for the immobilization of reptiles, most commonly crocodilians. In
small animals it also may be used, in conjunction with anaesthesia, to obtain muscular relaxation in all
types of surgery, for radiographic examination, and for manipulation.
Dosage and directions for use. Doses of 0,33-9,3 mg/kg have been used in crocodilians102,103. Lower
doses were required in alligators when diazepam (0,22-0,62 mg/kg) was given 20 minutes before the
suxamethonium103. A dose of 0,14-0,37 mg/kg suxamethonium produced immobilization in 5-15
minutes using this dosage regime.
Suxamethonium must be administered intravenously or intramuscularly. Absorption following
subcutaneous injection is too slow: the drug is metabolized by plasma pseudo-cholinesterase before it is
able to exert its effect. Intramuscular injection must be given in a large, superficial muscle, such as the
thigh or tail base, in crocodilians.
As with gallamine and other neuromuscular blocking agents, general or local anaesthesia must be given
before any painful procedures are performed.
Side-effects and special precautions. The side-effects of suxamethonium are associated with the initial
depolarization, and consist of uncoordinated muscular contractions, salivation, and sometimes severe
hypertension and bradycardia followed by tachycardia. The hypertension seems to be partially
mediated by the dyspnoea caused by suxamethonium. Depolarizing neuromuscular blocking agents
cause a release of potassium from skeletal muscle: elevation of serum potassium may therefore result,
particularly if repeated injections are given. Occasional occurrence of malignant hyperthermia in
humans has been attributed to suxamethonium, especially when used in conjunction with halothane.
Differences in the effects of suxamethonium can be potentiated by differences in levels or activity of
cholinesterase: such differences are observed between species, between young and old animals, due to
nutritional differences, or as result of recent exposure to drugs. Exposure to cholinesterase inhibitors
(such as organophosphate pesticides and anthelmintics, including exposure within the preceding month,
carbamates, and procaine) tends to increase the intensity and duration of action of suxamethonium.
Phenothiazine tranquillizers also have some anticholinesterase activity.
As with gallamine, the effects of suxamethonium may be enhanced by other drugs with neuromuscular
blocking effects.
Reversal of drug effects. Antagonists are not available, but are usually not necessary if sustained
artificial respiration is provided. The use of neostigmine is generally contra-indicated, particularly in
the initial stages following administration of suxamethonium: neostigmine, as with other cholinesterase
inhibitors, prolongs the duration of action and increases the intensity of the suxamethonium effect.
Toxicity and particulars of its treatment. Prolonged muscle paralysis, respiratory depression, and apnoea
occur with overdosage. Treatment is by positive pressure artificial respiration with oxygen and
maintenance of a patent airway until recovery of normal respiration is assured. Potential toxicity in
humans necessitates that this drug be used with the utmost caution.
Stability and storage instructions. Solutions of suxamethonium should always be refrigerated or kept on
ice in the field, since it undergoes spontaneous hydrolysis. Protect from light.
8 Drug antagonists
Until recently only a very few antidotes were available that could be used to reverse the pharmacological effects of
immobilizing drugs. Antidotes that have become available recently have increased the effectiveness and potential safety of
some of the immobilizing drugs and drug mixtures previously used without a specific antidote23. They are used in wild
animals, particularly under free-ranging conditions, to counter the effects of immobilization. Antidotes are useful for
ensuring that an immobilized animal regains its feet at the earliest opportunity, and also for the reversal of life-threatening
adverse reactions that occasionally occur. Drugs currently available to reverse the pharmacological effects of immobilizing
agents are summarized in Table 10.
Antidotes act either directly as pharmacological antagonists, or indirectly by physiological antagonism. A pharmacological
antagonist competes for the same receptor(s) as the agonist: depending on the relative agonist/antagonist concentration at
the receptor site, as well as the respective affinity for the receptor(s), the antagonist will displace the agonist and block
further agonistic effect. Pharmacological antagonists have no intrinsic pharmacological activity, although mixed
antagonists, such as some of the opioid antidotes, have both antagonistic and agonistic activity. These effects are not
generally displayed on the same receptor, but on different receptors for drugs with multireceptor action. For example,
mixed opioid antagonists reverse the immobilizing effects of opioids, but retain agonistic activity on the receptors
responsible for respiratory depression. Except for general anaesthetics and neuroleptics, specific pharmacological
antagonists are available for all other immobilizing drugs used in the wildlife field.
Physiological antagonists are drugs that cause opposing pharmacological effects to agonists by acting on different
receptors. These drugs are used particularly to reverse the cardiopulmonary depressant effects caused by some
immobilizing drugs, but may also promote recovery from the central nervous system depressant effects.
Reversal of the immobilizing effects caused by competitive, non-depolarizing, neuromuscular blockers is achieved by
agonistic effect. Cholinesterase inhibitors (e.g., neostigmine), which prevent the destruction of acetylcholine, increase the
endogenous neurotransmitter concentration at the receptor site and thus displace the neuromuscular blocker.
Pharmacological and physiological antagonists are listed as Schedule 4 substances and lower. No veterinary prescription is
required for drugs listed below Schedule 3, and these drugs can be obtained directly from a pharmacist.
8.1 Pharmacological antagonists
8.1.1 Opioid antagonists
Mixed and pure pharmacological opioid antagonists are available. Diprenorphine and nalorphine are
classified as mixed antagonists, since they have antagonistic as well as some inherent agonistic activity.
These drugs have been used successfully for many years to reverse the immobilizing effects of etorphine
and fentanyl in game capture. They retain some agonistic activity on the respiratory system35, and
should, therefore, be used with care in animals with severe respiratory depression. They should
preferably not be used in humans accidentally poisoned by opioids.
Naloxone is a specific opioid antagonist with no known agonistic activity. It is a very important drug for
emergency treatment of accidental opioid poisoning in humans. Its relatively short action, large dose
volume, and cost preclude its general use as an opioid antagonist in wild animals. Naltrexone and
nalmephene (a derivative of naltrexone) are newer pure opioid antagonists that are longer-acting and
which have a dose volume more suitable for general use. These drugs hold particular promise as
antagonists to the longer-acting opioid drugs, such as carfentanil, to prevent the occurrence of
'renarcotization'105. Nalmephene has only been used to a limited extent in wild animals60.
Diprenorphine hydrochloride (M5050®) and naloxone (Narcan®) are the only opioid antagonists
currently registered for veterinary use in South Africa.
8.1.1.1 Diprenorphine
Diprenorphine hydrochloride (M5050®, cyprenorphine, M-285, Revivon®)
(N-(cyclopropylmethyl)-6,7,8,14-tetrahydro-7-alpha-(1-hydroxy-1-methylethyl)-6,14-endo-ethano-noro
ripavine hydrochloride) is available as a 12 mg/ml injectable solution. It is supplied as M5050® in South
Africa by Krüger-Med Pharmaceuticals (Pty) Ltd in 10 ml vials filled to 5 ml, supplied together with
etorphine (M99®, Krüger-Med Pharmaceuticals) for use in game capture.
Chemical and physical characteristics. Diprenorphine hydrochloride is a white, or almost white,
crystalline powder that is soluble in 30 parts of water. A 2% m/v solution has a pH of 4,5-6,0.
Pharmacological action. Diprenorphine is a mixed opioid antagonist, retaining some agonistic activity.
Recovery times following opioid immobilization are within a few seconds to four minutes after
intravenous administration. Fentanyl immobilization is reversed more rapidly than etorphine
immobilization, while recovery is longer with carfentanil106. Intramuscular administration of
diprenorphine requires 5-20 minutes to reverse the CNS depressant effects of etorphine35.
Indications. Diprenorphine is used for the reversal of immobilization by opioids in domestic and
non-domestic animals.
Dosage and directions for use. Diprenorphine is administered intravenously or intramuscularly. Most
consistent results are obtained when an etorphine to diprenorphine ratio of 1:2 is used (i.e., 1 mg of
etorphine is antagonized by 2 mg of diprenorphine). However, the dosage varies according to mass of
the animal, and should be given according to the biological requirements of the particular animal.
Side-effects and special precautions. Due to some agonistic effect that is retained by diprenorphine,
respiratory depression caused by opioids may be aggravated, particularly in humans and following
repeated injection. Recycling of opioids resulting in 'renarcotization' has occasionally been observed
2-72 hours after immobilization60. The likelihood of recycling varies between species and subspecies,
type of opioid used, and type of antagonist used. Use of diprenorphine as an antagonist may contribute
to recycling of carfentanil, a longer acting opioid. Both etorphine and fentanyl can be stored and
secreted by the stomach60. With reabsorption of these drugs, there may be a depression of respiration in
apparently recovered patients as much as four hours after injection of short-acting antagonists. An
inadequate dose of diprenorphine is an important cause of recycling.
Toxicity and particulars of its treatment. Diprenorphine is a relatively safe product. Accidental injection
of the operator or overdosage of the animal is far less likely to cause toxic effects than the opioid.
Treatment is symptomatic and supportive. The use of alternative opioid antagonists is not
recommended.
Stability and storage instructions. Store in a cool place, below 25°C. Protect from light by keeping the
drug in the supplied container.
8.1.1.2 Nalorphine
Nalorphine hydrobromide (Lethidrone®, Nalorphine Injection®) and nalorphine hydrochloride
(Nalline®) are 20 mg/ml and 5 mg/ml injectable solutions, respectively. Nalorphine was one of the first
opioid antagonists developed, and has been used widely for many years. It is no longer marketed in
South Africa, but can be obtained as a dispensed medicine from Kyron Laboratories on veterinary
prescription for specified patients.
Nalorphine is a morphine derivative in which an N-methyl group has been replaced with a N-allyl
group. It acts as an antagonist at the mu-receptors, as partial agonist at kappa-receptors, and as an
agonist at sigma-receptors60. Under some circumstances, nalorphine was preferred to diprenorphine
because it produced a more thorough or complete reversal of narcosis107.
The effects following intravenous administration of nalorphine to the immobilized animal are almost
immediate. Within a few seconds there is a dramatic increase in respiration rate and minute volume.
Locomotion is usually restored after one minute. Nalorphine is preferably administered intravenously,
but may be given intramuscularly or subcutaneously. The average dosage rate for most species is 0,3-0,9
mg/kg.
In the absence of an opioid agonist, nalorphine produces CNS depression and analgesia as a result of its
partial agonist activity. Nalorphine may increase respiratory depressant effects of non-opioid CNS
depressants. Additional doses of nalorphine are contra-indicated when the initial reversal has failed35.
In these cases a pure antagonist should be used. A transient antagonist effect followed by renewed
depression may occur in some animals: this may be due to recycling of the opioid used, or to
underdosing23.
8.1.1.3 Naloxone
Naloxone hydrochloride (Narcan®, Boots) (17-allyl-4,5-alpha-epoxy3,14-dihydroxy-morphinan-6-one), is available as a 0,4 mg/ml injectable solution. It is supplied in a pack
of ten 1 ml ampoules.
Naloxone is a highly specific opioid antagonist with almost no agonistic properties, although some
side-effects have been reported in humans. In low doses it has a high affinity for mu-receptors. Large
doses (20-30 times that needed to block mu-receptors) are necessary for blockade of kappa-receptors,
and sigma-receptors are virtually insensitive to naloxone35. Non-specific arousal of the CNS, by for
example dopaminergic mechanisms and GABA antagonism, has been reported for naloxone35,24. This
non-opioid antagonistic effect may account for naloxone's reversal activity against diazepinones and
cyclohexylamines.
Naloxone is indicated mainly for reversal of the respiratory depressant effects of opioids, but has been
used widely for the reversal of all the immobilizing opioids. It has a potency of 10-30 times that of
nalorphine35. Its main drawback is related to its short half-life60. Apart from the traditional use as
antagonist to opioids and other CNS depressants it is also used clinically to treat various forms of shock
in animals35. A general dose rate of 0,04 mg/kg, given either intravenously or by intramuscular or
subcutaneous injection, is recommended for naloxone. A more reliable dosage regime involves two
injections, the first subcutaneously at 0,04-0,07 mg/kg, and the second intravenously or occasionally
intramuscularly at 0,10-0,25 mg/kg24. Naloxone is an essential component of the emergency kit that
should be present at all game capture operations.
High doses of naloxone can cause convulsions mimicking those produced by GABA antagonists.
Naloxone should be stored in a cool place under 25°C and protected from light.
8.1.1.4 Naltrexone and nalmephene
Naltrexone hydrochloride (Trexan®, DuPont-USA) and nalmephene (Nalmephene HCl®, Key
Pharmaceuticals) are specific opioid antagonists that are characterized by a long half-life in some
species, but not in all60. These products are currently not available for use in South Africa.
Naltrexone has been studied in a variety of species recently, but not in ruminants60,105. The drug is
reported to be 2,5 times as potent as naloxone108. An active metabolite of naltrexone, beta-naltrexol,
appears to play a role in the prolonged effect of the drug. In a recent study, naltrexone was the only
opioid antagonist that was fully effective in preventing recycling of carfentanil105. Naltrexone is
recommended at a ratio of at least 90:1 for reversal of carfentanil (90 part of naltrexone to 1 part of
carfentanil)60,105 and at a ratio of 20:1 for etorphine109.
Nalmephene is a naltrexone derivative that has only been tested to a limited extent in wild animals60. A
long-term effect in deer immobilized with etorphine was observed when nalmephene was given at a ratio
of 10 parts of nalmephene: 1 part of etorphine110.
8.1.2 Alpha-2-antagonists
The non-selective alpha-2-adrenoceptor antagonists tolazoline, phentolamine, yohimbine, and idazoxan,
have been used mainly to reverse the pharmacological effects of xylazine and detomidine. Tolazoline
and phentolamine have activity on both alpha-1- and alpha-2-adrenoceptors, whereas yohimbine and
idazoxan are more specific and have preferential affinity for the alpha-2-adrenoceptors, although
significant alpha-1-adrenoceptor binding does occur. Atipamezole is a potent alpha-2-adrenoceptor
antagonist with a high degree of selectivity and specificity for these receptors. Yohimbine is the only
alpha-2-adrenoceptor antagonist currently available in South Africa.
8.1.2.1 Yohimbine
Yohimbine hydrochloride is the hydrochloride of the indole-alkylamine alkaloid found in the bark of the
yohimbe tree (Pausinystalia yohimbe) and in the root of Rauwolfia sp. It can be obtained as a dispensed
medicine in injectable solutions of either 1,25 mg/ml in a 20 ml vial or 6,25 mg/ml in a 50 ml vial from
Kyron Laboratories on veterinary prescription for specified patients.
Yohimbine is a competitive antagonist that is selective for alpha-2-adrenergic receptors35. It is used as a
specific antagonist of alpha-2-adrenoceptor agonist sedatives, in particular xylazine. Additionally,
yohimbine antagonizes other CNS depressants that affect synaptic mechanisms, including barbiturates,
cyclohexylamines, and diazepinones, by binding on the same receptor sites, as well as on either the
alpha-2-adrenoceptors or secondary receptor sites (cholinergic, serotonergic and GABAergic)35.
Yohimbine is recommended for intravenous administration, but can also be given intramuscularly at a
dosage rate of 0,125-0,25 mg/kg1. Intravenous injection must be administered slowly111. The response
times of yohimbine are relatively short.
The safety margin of yohimbine appears to be relatively narrow: three out of four sheep that had
received xylazine at 0,4 mg/kg died within 2-3 minutes of receiving an intravenous injection of
yohimbine at 0,8 mg/kg111. Yohimbine readily penetrates the CNS, where it acts to increase blood
pressure and heart rate. Other side-effects include cardiac arrhythmias, anxiety, and manic reactions. It
is contra-indicated in renal or hepatic disease.
The concurrent use of yohimbine and 4-aminopyridine has been advocated for potentiation of reversal
of xylazine sedation and pentobarbitone anaesthesia. These drugs must not be combined in the same
syringe, but should be injected separately: they are chemically incompatible and precipitate, forming a
cloudy solution, when mixed.
8.1.2.2 Atipamezole
Atipamezole hydrochloride (Antisedan®, Ciba Geigy) (4-(2-ethyl
-2,3-dihydro-1H-inden-2-yl)-1H-imidazole hydrochloride) is available as a 5 mg/ml injectable solution
and is supplied in 10 ml multi-dose vials. It is currently in the process of registration for use in dogs and
cats in South Africa.
Atipamezole is a potent, selective, and specific alpha-adrenoceptor antagonist, exhibiting mainly
alpha-2-adrenoceptor activity at both central and peripheral receptor sites1,82. The alpha-2/alpha-1
selectivity ratio for atipamizole is 8526, compared to 27 and 40 for idazoxan and yohimbine,
respectively1. Atipamezole has no other significant receptor interaction. It is absorbed rapidly after
intramuscular injection (reaches maximum serum concentration after only 10 minutes in dogs) and has
an elimination half-life of 2,6 hours87.
Atipamezole has been developed specifically as an antagonist of medetomidine in animals112. It is able
to antagonize behavioural, cardiovascular, gastro-intestinal, neurochemical, and hypothermic effects of
medetomidine. Atipamezole has been used successfully in a wide range of non-domestic animals to
reverse the effects of medetomidine and xylazine38. The recommended dose of atipamezole is 4-5 times
the medetomidine dose in ruminants. Best results are obtained when 100-150 µg/kg is administered
intravenously and the rest subcutaneously. Because the required dose of cyclohexylamines (e.g.,
ketamine) is relatively high in carnivores, medetomidine and xylazine metabolism and excretion is
relatively advanced by the time the anaesthetic starts to wear off. A lower atipamezole dose of only 2-3
times the medetomidine dose is therefore recommended in carnivores. Intramuscular administration is
preferred. Reversal of medetomidine effects occurs within 5-10 minutes after intramuscular injection
and within two minutes when given intravenously. Recovery is smooth, and animals are generally able
to stand with the first attempt.
The dose of atipamezole should be reduced if it is administered late in the immobilization phase when a
large part of medetomidine has already been eliminated.
Xylazine-induced immobilization is rapidly reversed by 1 mg of atipamezole for every 8-12 mg of
xylazine used.
High doses of atipamezole may result in a transient nervousness or over-alertness1. Underdosing, or
intravenous administration alone, may result in re-sedation between 30 minutes and 4 hours after
reversal1.
Atipamezole must be stored below 25°C and must be protected from light.
8.1.2.3 Tolazoline and idazoxan
Tolazoline hydrochloride (Priscoline®, Ciba Geigy) is a non-selective alpha-adrenoceptor antagonist
that has been used for the reversal of xylazine effects. It has a wide range of pharmacological effects,
including adrenergic blocking and sympathomimetic, antihistaminic, and antihypertensive actions35.
Tolazoline is less potent than yohimbine as an alpha-2-adrenoceptor blocking agent. Tolazoline has been
used at doses of 0,5-5,0 mg/kg.
Idazoxan, an imidazoline derivative, is a potent alpha-2-adrenoceptor antagonist with a high degree of
selectivity and specificity for these receptors. It is up to 10 times more potent than yohimbine. Idazoxan
has been used to reverse sedation and respiratory and cardiovascular depression produced by xylazine
in calves and sheep. More rapid and complete reversal of xylazine immobilization was achieved with
idazoxan relative to yohimbine113, 114. Respiratory blood gas changes induced by detomidine and
xylazine were abolished by idazoxan in sheep115. The effective dose is still under investigation, but it
appears that 0,03-0,10 mg/kg is adequate for most purposes. RX821002A is a more potent analogue of
idazoxan.
None of these drugs are currently available in South Africa.
8.1.3 Diazepinone antagonists
The recent introduction of a diazepinone antagonist has opened the way for wider application of this
group of drugs in game capture. Flumazenil, a benzodiazepine antagonist, is able to reverse the
behavioural, neurological, and electrophysiological effects of all diazepinones78. A close analog to
flumazenil, RO 15-3505, has also been used to reverse the effects of diazepinones77. Flumazenil
(Anexate®) is available in South Africa as a human medicine and is not registered for veterinary use.
8.1.3.1 Flumazenil
Flumazenil (Anexate®, Roche) (ethyl
8-fluoro-5,6-dihydro-5-methyl-6-oxo-4H-imidazol[1,5-alpha][1,4]benzodiazepine-3-carboxylate),
previously known as flumazepil or RO-151788, is available as a 0,1 mg/ml injectable solution. It is
supplied in 5 ml and 10 ml ampoules.
Flumazenil is a benzodiazepine antagonist that acts competitively at CNS benzodiazepine receptors, but
is ineffective at peripheral benzodiazepine binding sites. It is indicated for the reversal of the central
effects of diazepinones in humans. The usual initial dose is 200 µg (total dose) given over 15 seconds,
followed at intervals of 60 seconds by further doses of 100 µg if required, to a maximum of 1 mg or
occasionally 2 mg. The usual dose range is 300-600 µg. Very few studies have been performed in
animals77. In wild animals, a dose of 1-2 mg/kg intramuscularly of RO 15-3505, an analogue of
flumazenil, has been used to reverse climazolam. In the case of flumazenil, a dose ratio of
climazolam:flumazenil of 10:1 has been recommended. Flumazenil has been described as devoid of
side-effects at recommended doses. At high doses, convulsions may occur.
8.1.4 Competitive peripheral muscle relaxant antidotes
8.1.4.1 Neostigmine
Neostigmine methylsulphate (Centaur Neostigmine Injection, Milborrow Animal Health; Prostigmin®,
Roche) is a synthetic analogue of the alkaloid physostigmine. It is available for veterinary use as a 2,5
mg/ml injectable solution in a 50 ml vial.
Neostigmine is an anticholinesterase substance that inhibits the enzymatic hydrolysis of acetylcholine by
acetylcholinesterases. With the inhibition of acetylcholinesterase, neuronally released acetylcholine
accumulates at the neuromuscular junction and competes with the non-depolarizing muscle relaxant,
gallamine, for the nicotinic receptors. The increased acetylcholine concentration is able to generate an
end-plate potential large enough to induce a propagated action potential, thereby effectively reversing
the muscle relaxant effects of gallamine.
Neostigmine also combines with cholinoceptors other than those of acetylcholine, and is able to exert
significant muscarinic action despite being to some extent selective for the neuromuscular junction 116.
Animals can be protected from the undesirable muscarinic side-effects by the initial administration of
atropine at 0,04 mg/kg followed by an intravenous injection of 0,025 mg/kg neostigmine.
Neostigmine can directly stimulate both pre- and post-junctional cholinoceptors. In the absence of a
non-depolarizing blocker, or with an overdose, prevention of the destruction of acetylcholine leads to an
accumulation of acetylcholine, thereby inducing a depolarizing blockade. For this reason, neostigmine is
contra-indicated as an antidote of the depolarizing peripheral muscle relaxant suxamethonium.
8.2 Physiological antagonists
8.2.1 Analeptics
Analeptics are drugs that are used to stimulate cardiopulmonary function. All analeptics physiologically
antagonize CNS depressants by acting as medullary stimulants. Their action is not confined only to the
medullary respiratory centre: they may also stimulate the vagal, vomition, and vasomotor centres, as
well as other cerebral functions, including consciousness. Analeptic drugs vary markedly in their total
pharmacological action35: some drugs, such as the methylxanthines, influence the function of the CNS
only secondarily, while primarily affecting another system in the body. A specific type of CNS stimulant
action may be elicited, such as the stimulation of the sensory areas of the brain, by methylxanthines,
whereas other drugs act directly or reflexly upon the respiratory centre to counteract respiratory
collapse (e.g., doxapram).
Drugs used for analeptic effect generally induce arousal characterized by a partial return of
consciousness. Animals do not return to a state of normal cerebral or locomotory function, but bump
and stagger around. The period of stimulation is normally very brief. A major drawback of analeptic
drugs is that the initial stimulation may give a false sense of security, temporarily relaxing operator
vigilance and/or application of other cardiopulmonary resuscitation measures. Also, if CNS stimulation
does occur and then subsides the secondary depression following stimulation could summate with the
depression produced by the injectable anaesthetic36. For most analeptics, a narrow margin exists
between respiratory and vasomotor stimulation and convulsant effects.
A number of analeptic drugs previously available (e.g., bemegride, nikethamide, and picrotoxin) have
become obsolete because of their doubtful value in chemical cardiopulmonary resuscitation due to their
potentially convulsogenic nature35. The best therapy for respiratory collapse consists of applying
artificial ventilation. Unfortunately, it is not always possible in veterinary practice, particularly under
field conditions and during game capture operations, to apply modern resuscitation measures with
oxygen equipment. There thus remains a need for, and a reliance on, analeptic drugs under these
conditions.
Doxapram is an important analeptic drug used frequently in wild animals. 4-Aminopyridine is
occasionally used as a physiological antagonist for xylazine, but may have a wider application. Both are
Schedule 4 substances.
8.2.1.1 Doxapram
Doxapram hydrochloride (Dopram®, Continental Ethicals) (1-ethyl-3,3-diphenyl-2-pyrrolidinone
hydrochloride) is available as a 20 mg/ml injectable solution supplied in a 20 ml bottle. It is available as a
human medicine in South Africa and is not registered for veterinary use.
Doxapram acts partly on the medullary respiratory centre, but primarily stimulates peripheral aortic
and carotid body chemoreceptors. The principal effect on ventilation is an increase in tidal volume. In
dogs, respiratory tidal volume is increased by 200% in one minute following administration of
doxapram35. Effects following a single injection of doxapram are maintained for approximately 4-6
minutes. Overall improvement in ventilation is reflected by changes in the acid-base status of the blood,
as well as in the oxygen tension of arterial blood. A vasopressor effect of doxapram occurs rapidly and
concurrently with respiratory effects. These pressor effects have been associated with activation of the
sympathetic nervous system, and occur only at doses not higher than 2 mg/kg: higher doses of up to 4
mg/kg given intravenously result in a transient hypotension. Differences in sensitivity to the effects of
doxapram occur between species, with the sheep and rabbit being the least sensitive35.
Doxapram is indicated for the reversal of respiratory depression caused by anaesthetic and opioid
drugs. Many wild animals are routinely administered doxapram immediately following immobilization
using etorphine. It has also been found useful for the reversal of cyclohexylamine CNS depression and
xylazine sedation. The usual dose of doxapram is 0,5-2,0 mg/kg. Higher doses of up to 11 mg/kg may be
required to reverse respiratory depression caused by barbiturates. Doses can be repeated after 15-20
minutes. Doxapram is administered either intravenously or subcutaneously. In life-threatening
respiratory arrest, doxapram is preferably given by slow intravenous infusion to effect, until the
breathing re-starts36. The injectable solution is incompatible with alkaline solutions, such as
aminophylline and thiopentone, and will precipitate upon mixture.
Unlike other analeptics, doxapram has a wide safety margin: the convulsant dose of doxapram is 70-75
times the dose that stimulates respiratory centre activity35. Its brief duration of action may result in
relapse of respiratory and CNS depression.
Doxapram hydrochloride injection should be stored below 25°C, but should not be allowed to freeze.
8.2.1.2 4-Aminopyridine
4-Aminopyridine (4-AP) is not readily available in South Africa, but can be obtained as a dispensed
medicine from Kyron Laboratories on veterinary prescription for specified patients. It is a CNS
stimulant and has considerable potential as an antagonist for a number of CNS depressants. Its main
indication has been for the reversal of the effects of xylazine, concurrently with
yohimbine117,118,119,120. However, in the past 4-aminopyridine was used clinically in humans as an
antagonist to d-tubocurarine.
4-Aminopyridine appears to act by facilitating neuronal uptake of Ca++ and enhancement of ACh
release at the neuromuscular junction35. It may also produce a selective blockade of K+ channels in
excitable membranes. The CNS effects of 4-aminopyridine are less well understood, but it has been
shown to reverse the effects of a number of CNS depressants.
The usual dosage of 4-aminopyridine is 0,25-0,50 mg/kg. Treatment can be repeated once if necessary.
In overdosage it is a convulsogenic agent. In non-sedated cattle it produces aggressive behaviour, muscle
tremors, hyperaesthesia, and vocalization, without convulsions118.
9 Ancilliary drugs
9.1 Drugs used for cardiopulmonary support
Cardiopulmonary arrest may occasionally occur following immobilization or the use of psychotropic agents.
These effects may result from, for example, overdosing, anaphylactoid reactions, exertion, vascular shock,
increased susceptibility due to species differences, disease, debilitation, or in very young or old animals. Apart
from physical resuscitation, it is generally necessary to administer emergency chemical treatment to revive these
animals. Drugs used under these circumstances include adrenaline or isoprenaline for cardiac stimulation,
clenbuterol and theophylline preparations for bronchodilation and respiratory support, and corticosteroids for
anaphylactoid and other types of shock reactions. Analeptic drugs, as already discussed, may also be indicated.
Cardiac arrhythmias may be a further complication during immobilization, resulting from the use of drugs or
from the physiological and somatic stressors accompanying immobilization, capture, or translocation.
Bradycardia and AV-blocks are known to be caused by some drugs, such as xylazine and other
alpha-2-adrenoceptor agonists. Cholinergic bradycardia may also result from a cholinergic response that
overrides the adrenergic alarm responses during immobilization. Tachyarrhythmias may occur as a result of the
use of immobilization drugs, such as the cyclohexylamines, or due to the capture and handling procedures, but
occur less frequently. The most commonly used drugs for the treatment of arrhythmias in animals are atropine
for bradyarrhythmias and lignocaine for tachyarrhythmias.
9.1.1 Adrenaline
Adrenaline (Centaur Adrenaline Injection®, Milborrow Animal Health; Adrenaline Injection®,
Labethica; Adrenaline Chloride®, Maybaker) is available as a 1 mg/ml (1:1000) injectable solution. It is
a sympathomimetic drug with mixed alpha and beta adrenoceptor activity resulting in an initial
increase in blood pressure and an immediate stimulation of cardiac function upon administration.
Adrenaline is usually reserved for acute cardiovascular collapse, cardiac arrest, and acute
anaphylactoid reactions. The drug is administered intravenously or intramuscularly at a dose of 1-3
µg/kg. Since intravenous injection may precipitate ventricular fibrillation it must be diluted by at least
1:10 in normal saline and injected with care when used by this route. With cardiac arrest, 0,5-1,0 ml of a
1:10000 solution may be administered directly into the left ventricular chamber or intratracheally in an
attempt to restore myocardial contraction. Larger doses may be required under certain circumstances.
Adrenaline must be administered with care to animals receiving anaesthetic agents (e.g., halothane) that
sensitize the myocardium to the effects of catecholamines.
9.1.2 Isoprenaline
Isoprenaline or isoproterenol hydrochloride (Imuprel®, Keatings; Isuprel®, Winthrop) is available as a
200 µg/ml (1:5000) injectable solution. It is a synthetic sympathomimetic structurally related to
adrenaline: it stimulates beta-adrenoceptors directly but has little effect on alpha-receptors.
Isoprenaline is indicated for the treatment of AV-block with syncope or severe bradyarrhythmia. For
emergency treatment 0,05-0,10 µg/kg isoprenaline is injected intravenously slowly over a period of a
minute, using a 1:50000 dilution. Treatment should be continued by slow intravenous drip or repeated
by intramuscular injection.
Isoproterenol is severely arrhythmogenic in equids, and should preferably not be used in this group.
9.1.3 Theophylline preparations
Aminophyline (Aminophyline®, Searle; Peterphylline®, Labethica) and etamiphylline camsylate
(Millophyline Injection®, Milborrow Animal Health) are theophylline preparations used primarily for
the treatment of bronchospasm. They are also capable of producing positive cardiac inotropic, diuretic
and CNS stimulatory effects. Slow intravenous injection of 6-15 mg/kg is recommended.
9.1.4 Clenbuterol
Clenbuterol hydrochloride (Planipart®, Janssen) is available as a 0,03 mg/ml injectable solution. It is a
synthetic beta-adrenoceptor agonist with predominantly beta-2 activity. It is indicated for the treatment
of acute bronchospasm at a dose of 0,8 µg/kg, intravenously. A transient, severe tachycardia may occur
following treatment.
9.1.5 Atropine
Atropine sulphate (Atropine Injection®, Milborrow Animal Health) is available as 0,5 mg and 10 mg/ml
injectable solutions. It is a parasympathomimetic drug that is indicated for the treatment or relief of
bradycardia and AV-block associated with the use of alpha-2-adrenoceptor agonists. Atropine may be
used as a premedication, when possible, or is injected immediately following immobilization. An
intravenous or intramuscular injection of 0,04 mg/kg is recommended. Because of prolonged mydriasis
and cycloplegia, atropine is not recommended for routine use in free-ranging wild animals.
9.1.6 Lignocaine
Lignocaine hydrochloride (Lignocaine Injection®, Milborrow Animal Health) is available as a 2%
injectable solution. It is a local anaesthetic recommended for the treatment of ventricular arrhythmias
in animals. Care must be taken not to use lignocaine solutions that contain adrenaline. An
intramuscular injection of 4-8 mg/kg is recommended. In equids the recommended dose is 0,5 mg/kg.
Severe convulsions may occur following intravenous injection.
9.2 Corticosteroids
High doses of intravenous glucocorticosteroids, such as prednisolone sodium succinate (Solu-Delta-Cortef®,
Upjohn) and betamethasone sodium phosphate (Betsolan Soluble Injection®, Milborrow Animal Health) are
indicated for the treatment of acute cases of shock. Fluid therapy must either accompany or precede
glucocorticoid administration. Doses of more than 10 times the normal recommended dose are used.
Intramuscular injection of the longer acting glucocorticoid prednisolone, betamethasone, or dexamethasone
esters are frequently given routinely following capture to reduce the harmful effects of stress.
9.3 Drugs used for the reduction of stress
Non-domestic animals are exposed to a tremendous amount of physiological and somatic stress during normal
capture and handling procedures, transportation, and adaptation to new environments. Physiological stress is
brought about by the sudden and violent alarm, fear, apprehension, anxiety, and frustration that occurs, and is
practically unavoidable, during these operations. Somatic fear might result from chronic disease states, injuries,
drug-induced effects, hunger, intense thirst, or extremes in ambient temperature. Injuries and mortalities of
many wild animals following capture or translocation are in many cases due to this stress.
Deleterious aspects of capture, handling, and translocation of wild animals can be modified or alleviated by
appropriate management procedures, and by the use of drugs. The use of long-acting tranquillizers has been
regarded as a major breakthrough in this respect (see section on long-acting tranquillizers). Alternative drugs
have recently become available that also have the potential to either modify or alleviate stress, or the effects of
stress, in wild animals. These drugs include beta-blockers (e.g., carazolol (Suacron®, Upjohn)) used for
cardiosympatholysis, Serotonin antagonistsserotonin S2 antagonists (e.g., Ketanserinketanserin and tameridone
(R51163, Janssen Pharmaceutica))121,122 for anxiolysis, and diazepinones (e.g., brotizolam (Mederantil®,
Janssen Pharmaceutica)) for appetite stimulation. Only a few studies have been performed with these drugs in
wild animals, but with promising results. Further research is needed with these products to elucidate their
potential benefits when used either alone or in combination with long-acting tranquillizers.
9.4 Synergists
A number of drugs (including tranquillizers, sedatives, hyaluronidase, and hyoscine) used in wild animals are
used for their synergistic ability to enhance the quality of immobilization of the particular immobilizing drug. The
main objective for the use of these drugs is to shorten induction time and thereby reduce the cost and/or risk to
the animal60.
HyaluronidaseHyaluronidase has been added to the immobilization drug or drug mixtures in the immobilization
of thick skinned wild animals, such as the black and white rhinoceros, African elephant, giraffe, and buffalo to
increase the rate of absorption of the drug(s) and to reduce induction time60,97. The drug improves absorption of
drugs by its action on intercellular 'cement' in the matrix of connective tissue and by increasing tissue
permeability123. An increase in absorptive tissue area of as much as 40% may be achieved using
hyaluronidase35. Hyaluronidase (Hyalase®, Fisons) is available as a injectable solution of 1500iu per ampoule.
Mixtures of 150 iu/ml are recommended. It is stable in solutions of etorphine and xylazine for at least 48 hours,
provided storage temperatures are maintained below 30°C. Hyaluronidase is unstable when dissolved with
phencyclidine, and must therefore only be mixed with this drug just prior use124.
Phenothiazine and butyrophenone tranquillizers, alpha-2-adrenoceptor agonists, and diazepinone sedatives
potentiate the effect of immobilizing drugs, resulting in smoother induction and fewer side-effects60. Smaller
doses of the immobilizing drug are thus required when these drugs are included in a drug mixture. A potential
problem in using these mixtures is that complete recovery of the animal is delayed: however, this may only be a
matter of dose60.
HyoscineHyoscine butylbromide (Buscopan®, Boehringer Ingelheim), also known as scopolamine butylbromide,
has also been used as a synergist in immobilizing mixtures for a variety of antelope species, and is still used in
rhinoceros. It is the l-isomer of hyoscyamine, and is more potent than atropine, which is d,l-hyoscyamine. These
are both parasympatholytic drugs that block muscarinic receptors at the neuro-effector junction. Hyoscine
butylbromide is more lipid soluble than atropine, and affects the CNS more readily. Hyoscine has a slight sedative
effect and, when combined with morphine, it produces analgesia and amnesia (referred to as twilight sleep) in
human patients35. It is favoured for use in wild animals by some operators because it induces a state of catalepsy,
photophobia, and mydriasis125. This facilitates loading of rhinos into crates following administration of the
antidote to the immobilizing drug. However, as a result of mydriasis as well as cycloplegia, the animal is partially
blind, which makes the drug inappropriate for animals that are to be released immediately. This partial blindness
is temporary, but may remain detectable for a few days. Large differences in susceptibility to the effects of these
drugs exist between species.
10 References
1. Jalanka, H.H. 1991. Medetomidine, medetomidine-ketamine combinations and atipamezole in nondomestic mammals: A
clinical, physiological and comparative study. Academic dissertation, Department of Clinical Sciences, College of Veterinary
Medicine, Helsinki, Finland.
2. Hupka. 1926. Ein Beitrag zur Narkose und Kastration der Raubtiere. Dtche. Tier.rzl. Wochenschr. 34:887-889.
3. Berge, E. 1938. Betubung von Zootieren. Zool. Garten (NF) 10:33-36.
4. Montgomery, G.G. & Hawkins, R.E. 1967. Diazepam baiting for capture of white-tailed deer. J. Wildl. Manage.
31:464-468.
5. Thomas, J.W., Robinson, R.M. & Marburger, R.G. 1967. Use of diazepam in the capture and handling of cervids. J.
Wildl. Manage. 31:686-692.
6. Done, S.H., Lees, P., Dansie, O. & Watkins, L.W. 1975. Sedation and restraint of fallow deer with diazepam. Br. vet. J.
131:545-548.
7. Meuleman, T., Port, T.H., Stanley, T.H. & Williard, K.F. 1984. Immobilization of elk and moose with carfentenil. J.
Wildl. Manage. 48:258-262.
8. Fowler, M.E. 1982. Delivery systems for chemical immobilization. In: L. Nielsen, J.C. Haigh, & M.E. Fowler, (eds),
Chemical Immobilization of North American Wildlife, pp. 18-45. The Wisconsin Humane Society, Inc., Milwaukee,
Wisconsin.
9. Fowler, M.E. (ed.) 1986. Zoo and Wild Animal Medicine, 2nd ed. W.B. Saunders Co., Philedelphia.
10. Severinghaus, C.W. 1950. Anesthetization of white-tailed deer. Cornell Vet. 40:275-282.
11. Larsen, L.H. 1963. Restraint and anaesthesia of wild animals in captivity. Australian Vet. J. 39:73-80.
12. Jones, D.M. 1984. Physical and chemical methods of capturing deer. Vet. Rec. 114:109-112.
13. Pienaar, U. de V. 1973. Darting and injection equipment and techniques. In: E. Young (ed.), The Capture and Care of
Wild Animals, pp. 7-13. Human & Rousseaux, Cape Town.
14. Hall, T.C., Taft, E.B., Baker, W.H. & Aub, J.C. 1953. A preliminary report on the use of Flaxedil to produce paralysis in
white-tailed deer. J. Wildl. Manage. 17:924-926.
15. Post, G. 1959. The use of curare-like drugs on elk (Wapiti). J. Wildl. Dis. 23:471-478.
16. Crockford, J.A., Hayes, F.A., Jenkins, J.H. & Feurt, S.D. 1957. Nicotine salicylate for capturing deer. J. Wildl. Manage.
21:213-220.
17. Rausch, R.A. & Ritcey, R.W. 1961. Narcosis of moose with nicotine. J. Wildl. Manage. 25:326-328.
18. Hawkins, R.E., Autry, D.C. & Klimstra, W.D. 1967. Comparison of methods used to capture white-tailed deer. J. Wildl.
Manage. 31:460-464.
19. Harthoorn, A.M. 1962. Capture of the white (square-lipped) rhinoceros, Ceratotherium simum simum (Burchell), with
the use of drug immobilization technique. Can. J. Comp. Med. 26:203-207.
20. Hofmeyer, C.F.B. 1973. Introduction. In: E. Young (ed.), The Capture and Care of Wild Animals, pp. 1-3. Human &
Rousseaux, Cape Town.
21. Harthoorn, A.M. 1971. The capture and restraint of wild animals. In: L.R. Soma (ed.), Textbook of Veterinary
Anesthesia, pp. 404-437. The Williams & Wilkins Company, Baltimore.
22. Beck, C.C. 1972. Chemical restraint of exotic species. J. Zoo Anim. Med. 3:3-66.
23. Harthoorn, A.M. 1973. Review of wildlife capture drugs in common use. In: E. Young (ed.). The Capture and Care of
Wild Animals, pp. 14-34. Human & Rousseaux, Cape Town.
24. Haigh, J.C. 1982. Mammalian immobilizing drugs: their pharmacology and effects. In: L. Nielsen, J.C. Haigh, & M.E.
Fowler, (eds), Chemical Immobilization of North American Wildlife. pp. 46-62. The Wisconsin Humane Society, Inc.,
Milwaukee, Wisconsin.
25. Hatlapa, H.H.M., Wiesner, H. (eds.) 1982. Die Praxis der Wildtierimmobilisation. Verlag Paul Parey, Hamburg.
26. Swan, G.E. & Naude, T.W. 1992. Basic Pharmacology - Student Notes. Faculty of Veterinary Science, University of
Pretoria.
27. Parker, J.B.R. & Haigh, J.C. 1982. Human exposure to immobilizing drugs. In: L. Nielsen, J.C. Haigh, & M.E. Fowler,
(eds), Chemical Immobilization of North American Wildlife. pp. 119-136. The Wisconsin Humane Society, Inc., Milwaukee,
Wisconsin.
28. Firn, S. 1973. Accidental poisoning by animal immobilizing agent. Lancet 95.
29. Harthoorn, A.M. 1976. The Chemical Capture of Animals. Bailliere Tindall, London.
30. Allsup, F.C. 1977. Accidental self injection. Vet. Rec. 100:499.
31. Goodrich, P.G.E. 1977. Accidental self injection. Vet. Rec. 100:458-459.
32. Orr, C.M. 1977. Accidental self injection. Vet. Rec. 100:574.
33. Carruthers. D. et al. 1979. Xylazine hydrochloride (Rompun®) overdose in man. Clin. Toxicol. 15:281-285.
34. Sawyer, D.C., Hoogstraten, S. 1980. A ketamine experience - unintentional injection of ketamine hydrochloride into a
human. J.A.A.H.A. 16.
35. Booth, N.H. & McDonald, L.E. (Eds.), Veterinary Pharmacology and Therapeutics, 6th ed. Iowa State University Press.
36. Lees, P. 1991. Drugs acting on the central nervous system. In: G.C. Brander, D.M. Pugh, R.L. Bywater & W.L., Jenkins
(eds), Veterinary Applied Pharmacology & Therapeutics, 5th ed, pp. 315-391. Bailiere Tindall, London.
37. Boever, W.J., Holden, J., Kane, K.K. 1977. Use of Telazol TM (Cl-744) for chemical restraint and anaesthesia in wild and
exotic carnivores. VM/SAC, Exotic species, 1722-1725.
38. Jalanka, H.H. & Roeken, B.O. 1990. The use of medetomidine, medetomidine-ketamine combinations, and atipamezole
in nondomestic mammals: A review. Jnl. Zoo Wildl. Med. 21:259-282.
39. Muir, W.W. & Hubell, J.A.E. 1988. Cardiopulmonary and anesthetic effect of ketamine and its enantiomers. Am. J. Vet.
Res. 49:530-534.
40. Delatour, P., Jaussaud, P., Courtot, D., Fau, D. 1991. Enantioselective N-demethylation of ketamine in the horse. J. vet.
Pharmacol. Therap. 14:209-212.
41. Wright, M. 1982. Pharmacological effects of ketamine and its use in veterinary medicine JAVMA 180:1462-1471.
42. Gray, C.W., Bush, M & Beck, C.C. 1974. Clinical experience using C1744 in chemical restraint of exotic specimens. J.
Zoo. Ani. Med. 5:12-21.
43. Weisner, H. 1975. Zur Neuroleptanalgesie bei Zootesen und Gatterwild unter Anwendung des Tele-inject Systems.
Kleintier Praxis 20:18-24.
44. Davy, C.W., Trenenery, P.N., Edmunds, J.G., Altman, J.F.B. & Eichler, D.A. 1987. Local myotoxicity of ketamine in the
marmoset. Lab. Anim. 21:60-67.
45. Allen, J.L. 1986. Use of tolazoline as an antagonist to xylazine-ketamine induced immobilization in African elephants.
Am. J. Vet. Res. 47:781-783.
46. Allen, J.L. & Oosterhuis, J.E. 1986. Effect of tolazoline on xylazine-ketamine induced anesthesia in turkey vultures.
JAVMA 189:1011-1012.
47. Seal, U.S., Armstrong, D.L. & Simmons, L.G. 1987. Yohimbine hydrochloride reversal of ketamine hydrochloride and
xylazine hydrochloride immobilization in Bengal tigers and effects on hematology and serum chemistries. J. Wildl. Dis.
23:296-299.
48. King, J.M., Bertram, B.C.R. & Hamilton, P.H. 1977. Tiletamine and Zolazepam for immobilization of wild lions and
leopards. JAVMA 171:88-92.
49. Bush, M., Custer, R., Smeller, J., Bush, L.M., Seal, U.S. & Barton, R. 1978. The acid-base status of lions, Panthero leo,
immobilized with four drug combinations. J. Wildl. Dis. 14:102-109.
50. Bush, M. & Custer, R.S. 1980. Use of dissociative anesthetic for the immobilization of captive bears: blood gas,
hematology and biochemistry values. J. Wildl. Dis. 16:481-489.
51. Baker, J.R., Fedak, M.A., Anderson, S.S., Arnbom, T. & Baker, R. 1990. Use of a tiletamine-zolazepam mixture to
immobilise wild grey seals and southern elephant seals. Vet. Rec. 126:75-77.
52. Van Heerden, J., Burroughs, R.E.J., Dauth, J. & Dreyer, M.J. 1991. Immobilization of wild dogs (Lycaon pictus) with a
tiletamine hydrochloride/zolazepam hydrochloride combination and subsequent evaluation of selected blood chemistry
parameters. J. Wildl. Dis. 27:225-229.
53. Donham, K.J. & Rubino, M.J. 1977. Oral administration of phencyclidine in chimpanzees. Lab Animal:44-45.
54. Button, C., Meltzer, D.G.A. & Mülders, M.S.G. 1981. The electrocardiogram of the cheetah (Acinonyx jubatus). J. S.A.
vet Assoc. 52:233-235.
55. Button, C., Meltzer, D.G.A. & Mülders, M.S.G. 1981. Saffan induced poikilothermia in cheetah (Acinonyx jubatus). J.
S.A. vet Assoc. 52:237-238.
56. Jaffe, J.H. & Martin, W.R. 1990. Opioid analgesics and antagonists. In: A.G. Gilman, T.W. Rall, A.S. Nies & P. Taylor
(eds), The Pharmacological Basis of Therapeutics, 8th ed., pp. 485-521. Pergamon Press.
57. Akil, H., Watson, S.J., Young, E., Lewis, M.E., Khachaturian, H. & Walker J.M. 1984. Endogenous opioids: biology
and function. Annu. Rev. Neurosci. 7:223-255.
58. Simon, E.J. 1977. In: J.R. Smythies & R.J. Bradley (eds)., Receptors in Pharmacology, p 257. Marcel Dekker, New York.
59. Kania, B.F. 1985. Presynaptic stimulation of dopaminergic CNS structures in sheep as a mechanism of immobilising
action of Immobyl (fentanyl + azaperone). Res. Vet. Sc. 38:179-183.
60. Haigh, J.C. 1990. Opioids in zoological medicine. J. Zoo Wildl. Med. 21:391-413.
61. Alford, B.T., Burkhart, R.L. & Johnson, W.P. 1974. Etorphine and diprenorphine as immobilizing and reversing agents
in captive and free-ranging mammals. J. Am. Vet. Med. Assoc. 164:702-705.
62. Van Jaarsveld, A.S., McKenzie, A.A., & Melzer, D.G.A. 1984. Chemical immobilization and anaesthesia of spotted
hyaenas Crocuta crocuta. S. Afr. J. Wildl. Res. 14:120-122.
63. Ebedes, H. 1989. Capturing by chemical (or drug) immobilisation. In: J. du P. Bothma (ed.), Game Ranch Management,
pp.415-422. Van Schaik, Pretoria.
64. Kock, R.A. & Pearce, P.C. 1985. Anaesthesia in zoo ungulates. J. Assoc. Vet. Anaesth. (U.K.) 13:59-88.
65. LeBlanc, P.H., Eicker, S.W., Curtis, M. & Beehler, B. 1987. Hypertension following etorphine anesthesia in a
rhinoceros (Diceros simus). Jnl. Zoo An. Med. 18:141-143.
66. Lumb, W.V. & Jones, E.W. 1984. Preanesthetic agents. In: V.W. Lumb & E.W. Jones (eds), Veterinary Anesthesia, 2nd
ed., pp 165-197. Lea & Febiger, Philadelphia.
67. Haigh, J.C. 1976. Fentanyl-based mixtures in exotic animal neurolept-analgesia. Ann. Proc., AAZV Congress, St. Louis.
pp. 164-180. H3
68. Reynolds, J.E.F., Parfit, K., Parsons, A.V. & Sweetman, S.C. 1989. Opioid analgesics. In: Martindale: The Extra
Pharmacopoeia, 29th ed., pp. 1294-1321, Pharmaceutical Press, London.
69. Keep, M.E. 1973. The use of etorphine hydrochloride (M99) (Reckitt), fentanyl (Janssen) and hyoscine hydrobromide
combination for field capture of white rhinoceros. Lammergeyer 19:28-30.
70. Soma, L.R. & Shields, D.R. 1964. Neuroleptanalgesia produced by fentanyl and droperidol. JAVMA 145:879-902.
71. De Vos, V. 1978. Immobilization of free-living wild animals using a new drug. Vet. Rec. 103:64-68.
72. Kreeger, T.J. & Seal, U.S. 1990. Immobilization of Gray Wolves (Canis lupus) with sufentanil citrate. J. Wildl. Dis.
26:561-563.
73. Moore, R.A., Yang, S.S. & McNicholas, K.W. 1985. Hemodynamic and anesthetic effects of sufentanil as the sole
anesthetic for pediatric cardiovascular surgery. Anesthesiology 65:355-358.
74. Stanley, T.H.S. & McJames 1986. Chemical immobilization using new high potency opioids and other drugs and drug
combinations with high therapeutic indices. U.S. Dep. Defence. Final rep. Contract DAAK11-84-K-0002, Washington, D.C.
40pp.
75. Stanley, T.H.S., McJames, S., Kimball, J., Port, J.D. & Pace, N.L. 1988. Immobilisation of elk with A3080. J. Wildl.
Manage. 52:577-581.
76. Reeves, J.G., Fragen, R.J., Vinik, R. & Greenblatt, D.J. 1985. Midazolam: Pharmacology and uses. Anaesthesiology
62:310-324.
77. Gutzwiller, Von A., Völlm, J. & Hamza, B. 1984. Einsatz des Benzodiazepins Climazolam bei Zoo- und Wildtieren.
Kleintier Praxis 29:319-332.
78. O'Sullivan, G.F. & Wade, D.N. 1987. Flumazenil in the management of acute drug overdosage with benzodiazepines
and other agents. Clin. Pharmacol. Ther. 42:254-259.
79. Scheinin, M. & MacDonald, E. 1989. An introduction to the pharmacology of alpha-2-adrenoceptors in the central
nervous system. Acta Vet. Scand. Suppl. 85:11-19.
80. Byland, D.B. 1985. Heterogeneity of alpha-2-adrenergic receptors. Pharmacol. Biochem. Behav. 22:835-843.
81. Murphy, T.J. & Byland, D.B. 1988. Characterization of alpha-2-adrenergic receptors in the OK cell, an opossum kidney
cell. J. Pharmacol. Exp. Ther. 244:571-578.
82. Vähä-Vähä, T. 1991. Pharmacological restraint - reversal in dogs and cats using medetomidine and atipamezole. Academic
dissertation, Department of Clinical Sciences, College of Veterinary Medicine, Helsinki, Finland. pp. 1-51.
83. Knight, A.P. 1980. Xylazine. J. Am. Vet. Med. Assoc. 176:454-455.
84. Virtanen, R. 1986. Pharmacology of detomidine and other alpha-2-adrenoceptor agonists in the brain. Acta. vet. Scand.
82:35-46.
85. Salonen, J.S. 1986. Pharmacokinetics of detomidine. Acta Vet. Scand. 82:59-66.
86. Hyman, W.B. 1992. Veterinary restraint drugs with special reference to detomidine. In: H. Ebedes (ed.), The Use of
Tranquillizers in Wildlife, pp. 25-30. Department of Agricultural Development, Pretoria.
87. Salonen, J.S. 1989. Pharmacokinetics of medetomidine. Acta Vet. Scand. 85:49-54.
88. Van Heerden, J., Swan, G.E., Dauth, J., Burroughs, R.E.J. & Dreyer, M.J. 1991. Sedation and immobilization of wild
dogs Lycaon pictus using medetomidine or a medetomidine-ketamine hydrochloride combination. S. Afr. J. Wildl. Res.
21:88-93.
89. Baldessarini, R.J. 1990. Drugs and the treatment of psychiatric disorders. In: A.G. Gilman, T.W. Rall, A.S. Nies & P.
Taylor (eds), The Pharmacological Basis of Therapeutics 8th ed, pp. 383-435. Pergamon Press. .
90. Lees, G.M. 1981. A hitch-hiker's guide to the galaxy of adrenoceptors. Brit. med. J. 283:173-178.
91. Tranquilli, W. & Thurmon, J.C. 1984. Alpha adrenoceptor pharmacology. J. Am. vet. Ass. 184:1400-1402.
92. Ruskoaho, H. 1986. Subtypes and functions of alpha-adrenoceptors. Acta vet. Scand. 82:17-28.
93. Colly, L.P. 1992. Azaperone - a safe and well-tried tranquillizer. In: H. Ebedes (ed.), The Use of Tranquillizers in Wildlife, pp.
21-22. Department of Agricultural Development, Pretoria.
94. Ebedes, H. & Burroughs, R.E.J. 1992. Long-acting neuroleptics in wildlife. In: H. Ebedes (ed.), The Use of Tranquillizers in
Wildlife, pp. 31-37. Department of Agricultural Development, Pretoria.
95. Lingjaerde, O. 1973. Some pharmacological aspects of depot neuroleptics. Acta Psych. Scand. 246(suppl.):9-14.
96. Hofmeyr, J.M. 1981. The use of haloperidol as a long-acting neuroleptic in game capture operations. J. S.A. vet. Assoc.
52:273-282.
97. Morkel, P. 1989. Drugs and dosages for capture and treatment of black rhinoceros (Diceros bicornis) in Namibia. Koedoe
32:65-68.
98. Ebedes, H. 1992. A note on haloperidol for translocation. In: H. Ebedes (ed.), The use of tranquilizers in wildlife, pp. 23-24.
Department of Agricultural Development, Pretoria.
99. Hofmeyr, J.M., Luchtenstein, H.G. & Mostert, P.K.N. 1977. Capture, handling and transport of springbok and the application of
long acting neuroleptics. Madoqua 10:123-130.
100. Taylor, P. 1990 Agents acting at the neuromuscular junction and autonomic ganglia. In: A.G. Gilman, T.W. Rall, A.S. Nies &
P. Taylor (eds), The Pharmacological Basis of Therapeutics, 8th ed., pp 166-186, Pergamon Press.
101. Button, C., Bertschinger, H.J. & Mülders, M.S.G. 1981. Haemodynamic and neurological responses of ventilated and apnoeic
calves to succinyldicholine. J. S.A. vet. Assoc. 52:283-288.
102. Loveridge, J.P. 1979. The immobilisation and anaesthesia of crocodilians. Int. Zoo Yb. 19:103-111.
103. Bennett, R.A. 1991. A review of anesthesia and chemical restraint in reptiles. J. Zoo Wildl. Med. 22:282-303.
104. Mayrhofer, O.K. 1952. Self experiments with succinylcholine chloride. Br. Med. J. 1:1332-1334.
105. Allen, J.L. 1989. Renarcotisation following carfentanil immobilization of nondomestic ungulates. J. Zoo Wildl. Med.
20:243-426.
106. Haigh, J.C. 1991. Immobilization of wapiti with carfentanil and xylazine and antagonism with diprenorphine, naloxone, and
naltrexone. J. Zoo Wildl. Med. 22:318-323.
107. Hofmeyr, J.M. 1974. Developments in the capture and airlift of roan antelope Hippotragus equinus equinus under narcosis to
the Etosha National Park. Madoqua 8:37-48.
108. Gonzales, J.P. & Brogden, R.N. 1988. Naltrexone: a review of its pharmacodynamic and pharmacokinetic properties and
therapeutic efficacy in the management of opioid dependence. Drugs 35:192-213.
109. Allen, J.L. 1990. Renarcotization following immobilisation of non-domestic equidae. J. Zoo Wildl. Med. 21:292-294.
110. Kreeger, T.J., Plotka, E.D. & Seal, U.S. 1987. Immobilization of white-tailed deer by etorphine and xylazine and its
antagonism by nalmephene and yohimbine. J. Wildl. Dis. 23:619-624.
111. Hsu, W.H., Schaffer, D.D. & Hanson, C.H. 1987. Effects of tolazoline and yohimbine on xylazine-induced central nervous
system depression, bradycardia, and tachypnea in sheep. J. Am. Vet. Med. Assoc. 190:423-426.
112. Karjalainen, A.J., Virtanen, R.E. & Karjalainen, A.L. 1986. Syntheses and effects of modification of the
4(5)-(2,3-dihydro-1H-inden-2-yl)imidazole ring system on alpha-adrenoceptor blocking activity. In: Abstracts of the IXth
International Symposium on Medical Chemistry. Berlin (West): Fachgruppe 'Medizinische Chemie' of Gesellschaft Deutcher
Chemiker, pp 268.
113. Crawshaw, G.J., Mehren, K.G. & Black, S. 1986. Antagonionism of xylazine and ketamine/xylazine combinations in exotic
species by idazoxan and RX821002A. Annu. Proc. Am. Assoc. Zoo Vet. pp. 1-2.
114. Kock, R.A., Harwood, J.P.P., Pearce, P.C. & Cinderey, R.N. 1989. The use of two novel alpha-2-adrenoceptor antagonists and
its analogue RX821002A in zoo and wild animals. J. Assoc. Vet. Anaesth. (U.K.) 16:4-10.
115. Waterman, A.E., Nolan, A. & Livingstone, A. 1987. Influence of idazoxan on the respiratory blood gas changes induced by
alpha-2-adrenoceptor agonist drugs in conscious sheep. Vet. Rec. 121:105-107.
116. Pugh, D.M. 1991. Local anaesthesia and voluntary-muscle relaxation. In: G.C. Brander, D.M. Pugh, R.L. Bywater & W.L.
Jenkins (eds), Veterinary Applied Pharmacology and Therapeutics, 5th ed, pp. 81-96. Bailliere Tindall, London.
117. Hatch, R.C., Booth, N.H., Clark, J.D., Crawford, L.M., Kitzman, J.V. & Wallner, B. 1982. Antagonism of xylazine by
4-aminopyridine and yohimbine. Am. J. Vet. Res. 43:1009-1014.
118. Kitzman, J.V., Booth, N.H., Hatch, R.C. & Wallner, B. 1982. Antagonism of xylazine sedation by 4-aminopyridine and
yohimbine in cattle. Am. J. Vet. Res. 43:2166-2169.
119. Komulainen, A. & Olson, M.E. 1991. Antagonism of ketamine-xylazine anesthesia in rats by administration of yohimbine,
tolazoline, or 4-aminopyridine. Am. J. Vet. Res. 52:585-588.
120. Verstegen, J., Fargetton, X., Zanker, S., Donnay, I. & Ectors, F. 1991. Antagonistic activities of atipamizole, 4-aminopyridine
and yohimbine against medetomidine/ketamine-induced anaesthesia in cats. Vet. Rec. 128:57-60.
121. Ooms, L.A.A. & Verheyen, A.K. 1982. Malignant hyperthermia: etiology, pathophysiology, and prevention. In: F. De Clerck
& P.M. Vanhoutte (eds), 5-Hydroxytryptamine in Peripheral Reactions, pp. 129-139. Raven Press, New York.
122. Degryse, A-D.A.Y. & Ooms, L.A.A. 1986. Comparative studies on cardiovascular, respiratory and gastro-intestinal effects of
the sedatives R51163 and xylazine in cattle. Drug Dev. Res. 8: 433-441.
123. Morton, D.J. & Kock, M.D. 1991. Stability of hyaluronidase in solution with etorphine and xylazine. Jnl. Zoo Wildl. Med.
22:345-347.
124. Foster, P.A. 1973. Immobilisation and anaesthesia of primates. In: E. Young (ed.), The Capture and Care of Wild Animals,
pp. 69-76. Human & Rousseaux, Cape Town.
125. Pienaar U. de V. 1973. The drug immobilisation of antelope species. In: E. Young (ed.), The Capture and Care of Wild
Animals, pp. 35-50. Human & Rousseaux, Cape Town.
Personal communications: Dr P. Morkel, Wildlife Veterinary Services, P O Box 80371, Olympia, Windhoek, Namibia; Dr H.
Ebedes, Department of Agricultural Development, P Bag X180, Pretoria, 0001 RSA.
If you have no navigation at the top of the page, go HERE.
Brought to you by www.wildnetafrica.com
© WildNet Africa (Pty.) Ltd. - Africa's Wildest Web
Disclaimer: the information on this page is used entirely at the reader's discretion, and is made available on
the express condition that no liability, expressed or implied, is accepted by WildNet Africa or any of its
associates, employees or subsidiaries for the accuracy, content or use thereof.