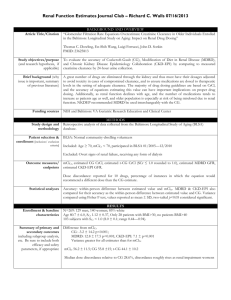
Summary of the used interacting drug physiologically
advertisement

Supplementary materials: Clinical Pharmacokinetics Title: Physiologically-based pharmacokinetic modeling of macitentan: prediction of drugdrug interactions Authors: Ruben de Kanter, Patricia N. Sidharta, Stéphane Delahaye, Carmela Gnerre, Jerome Segrestaa, Stephan Buchmann, Christopher Kohl and Alexander Treiber Corresponding Author: Ruben de Kanter Preclinical Pharmacokinetics and Metabolism Actelion Pharmaceuticals Ltd. Gewerbestrasse 16 CH-4123 Allschwil Switzerland phone: +41 61 565 5618 e-mail: ruben.de-kanter@actelion.com Details on experimental in vitro procedures Physical-chemical properties: The distribution-coefficient logD was determined using a slightly modified shake flask method which is based on a OECD guideline [1]. Briefly, the test compound was dissolved at 4 mM in the organic phase (n-octanol), which was further mixed with the aqueous buffered phase (67 mM phosphate buffer pH 7.4). After shaking overnight, the two phases were separated by centrifugation and the concentration of the test compound in each phase was measured by HPLC-UV. D is given by the direct quotient of the compound concentration in the organic and aqueous phases. The ionization constant pKa was determined with a multiwavelength spectrophotometric method, using the GLpKa / D-PAS (macitentan) or Sirius T3 (ACT-132577) instrument from Sirius Analytical (Forest Row, UK) [2]. Plasma protein binding: The fraction unbound in human plasma was determined using a Spectrum equilibrium dialyzer (Spectrum Chemicals & Laboratory Products) [3]. Pooled plasma was spiked with 100 - 300,000 ng/mL 14C-labeled macitentan or ACT-132577 and dialyzed against 0.02 M phosphate-buffered saline (pH 7.4) at 37 °C for 6 hours using a filter membrane with a molecular weight cut-off of 12-14 kDa. Aliquots taken from both donor and buffer compartments were analyzed by liquid scintillation counting and the fraction unbound was calculated as described [3]. P450 inhibition: P450 inhibition was studied using selective marker reactions as specified below. Substrates were used at a single concentration around their Km (Michaelis-Menten constant) and both macitentan and ACT-132577 were incubated at eight different concentrations, up to 50 µM. Solvent (negative) controls and well-known inhibitors (positive) controls were included. Microsomal incubations were initiated by the addition of NADPH-regenerating system and terminated by adding an excess of ice-cold organic solvent. A summary of experimental conditions is given in the table below. P450 isoform 1A2 2A6 2B6 2C8 2C9 2C19 2D6 2E1 3A4 3A4 marker reaction enzyme concentration, system and incubation period phenacetin-Odeethylation coumarin 7hydroxylation (S)-mephenytoin N-desmethylation paclitaxel 6-hydroxylation diclofenac 4’hydroxylation (S)-mephenytoin 4’-hydroxylation dextrometorphanO-demethylation chlorzoxazone 6hydroxylation midazolam 1’hydroxylation testosterone 6hydroxylation 1 mg/mL HLM, 30 min marker substrate concentration (~Km) 50 µM analytical method 0.025 mg/mL HLM, 6 min 1 µM HPLC-UV 200 pmol/mL recombinant CYP2B6, 40 min 0.25 mg/mL HLM, 20 min 50 µM 8 µM HPLC-radioactivity detection LC-MS/MS 0.1 mg/mL HLM, 6 min 5 µM LC-MS/MS 50 pmol/mL recombinant CYP2C19, 40 min 0.2 mg/mL HLM, 30 min 50 µM 8 µM HPLC-radioactivity detection LC-MS/MS 0.35 mg/mL HLM, 20 min 50 µM HPLC-UV 0.25 mg/mL HLM, 5 min 5 µM LC-MS/MS 0.3 mg/mL HLM, 7 min 40 µM HPLC-radioactivity detection or LCMS/MS LC-MS/MS HLM, human liver microsomes (pooled from 51 donors; Becton Dickinson, Basel, Switzerland); recombinant enzymes, SupersomesTM (BD Biosciences, Allschwil, Switzerland). HPLC-UV High-Pressure Liquid Chromatography with UV Detector; LCMS/MS Liquid chromatography-tandem mass spectrometry. The Ki values for CYP2C9 inhibition of both macitentan and ACT-132577, as well as Ki value for CYP3A4 inhibition of ACT-132577, were determined using four different substrate concentrations, between 2-fold below to 10-fold above the Km. Inhibitor concentrations were the same as used in the IC50 experiments. IC50 and Ki values were calculated by fitting the rate of metabolite production against inhibitor concentration using the transformed Michaelis-Menten equation. Time dependent inhibition was studied with or without a 30 min pre-incubation period in the presence of NADPH but without the probe substrate. At the end of the pre-incubation period, probe substrates were added at a single concentration of about 10 times the respective Km values, as indicated the table above. No shift in IC50 values was observed for any of the P450 enzymes studies (CYP2C9, CYP2D6 and CYP3A4). 2 Identification of P450 enzymes involved in macitentan and ACT-132577 metabolism: 14 C-labeled macitentan and ACT-132577 were incubated at 10 µM with human liver microsomes in the absence or presence of P450 enzyme-selective chemical inhibitors at concentrations 10 times their respective Ki's, as specified in the table below, or with recombinant enzymes in SupersomesTM (BD Biosciences, Allschwil, Switzerland) or BactosomesTM (Cypex, Dundee, UK). Pooled human liver microsomes (Becton Dickinson, Basel, Switzerland) at a protein concentration of 1 mg/mL or recombinant P450 enzymes at 100 pmol/mL were used in 0.1 M phosphate buffer (pH 7.4). Incubations were initiated by the addition of a NADPH-regenerating system and continued for 40 min at 37 °C, in a thermostatic shaker. Incubations were terminated by the addition of ice-cold methanol and samples were analyzed after protein precipitation by HPLC with radiodetection. P450 enzyme inhibitor concentration CYP2C8 CYP2C9 CYP2C19 CYP3A4 montelukast sulfaphenazole S-(+)-N-3-benzylnirvanol ketoconazole 5 µM 5 µM 5 µM 1 µM Details on dosing regimens: Dosing regimens in the simulations were matched to the referenced studies and were performed in the fed state, except the interaction study with rifampicin, which was done under fasted conditions [4]. Concomitantly dosed drugs were given at the same time, e.g. daily macitentan was dosed together with the first daily dose of twice daily rifampicin. Modeling was done assuming 250 mL fluid intake with dosing. Once a day ketoconazole interaction: 400 mg ketoconazole was dosed once daily from day 1 until day 24. Concomitantly, 10 mg macitentan was dosed once on day 5 [5]. Twice a day ketoconazole interaction: 200 mg ketoconazole was dosed twice daily from day 1 until day 24. Concomitantly, 10 mg macitentan was dosed once on day 5. Rifampicin interaction: subjects received a loading dose of 30 mg macitentan on the first day, followed by 10 mg macitentan once daily for 12 days. Concomitantly, 600 mg rifampicin was dosed daily from day 6 to day 12 [4]. Cyclosporine interaction: subjects received a loading dose of 30 mg macitentan on the first day followed by 10 mg macitentan once daily for 16 days. Concomitantly, 100 mg rifampicin was dosed twice daily from day 6 to day 12 [4]. Sildenafil interaction: subjects received a loading dose of 30 mg macitentan on the first day followed by 10 mg macitentan once daily for 3 days. Sildenafil was concomitantly dosed at 20 mg three times daily for 3 days and once on day 4 [6]. Warfarin interaction: subjects received a loading dose of 30 mg macitentan on the first day followed by 10 mg macitentan once daily for 8 days. On day 4, warfarin was given concomitantly as a single dose of 25 mg [7]. The drug interaction studies with erythromycin, itraconazole, ritonavir, diltiazem, verapamil, clarithromycin, saquinavir, phenytoin and carbamazepine were simulated using the dosing 3 scheme of the perpetrator drug presented in Table 9, for 24 days and a single concomitant dose of 10 mg macitentan on day 5. Details on the model population: Mean demographic data and scaling factors of the simulations (n=100 healthy male volunteers) geometric mean C.V. 27 25 % weight (kg) 80 17 % cardiac output (L/h) 365 10 % liver weight (g) 1711 17 % age (years) hepatocelularity (million cells / g liver) 110 22 % CYP3A4 liver content (pmol) 8573017 55 % CYP3A4 gut content (pmol) 60372 54 % C.V. coefficient of variation Geometric mean predicted tissue-partition coefficients for healthy subjects (n=100): macitentan ACT132577 Adipose Bone Brain Gut Heart Kidney Liver Lung Muscle Skin Spleen 0.45 0.32 0.25 0.31 0.20 0.20 0.18 0.23 0.34 0.37 0.17 0.16 0.11 0.06 0.17 0.16 0.14 0.09 0.21 0.28 0.28 0.10 4 Summary of the used interacting drug physiologically-based pharmacokinetic models - physical chemistry, absorption, distribution and interaction ketoco nazole rifampi cin cyclosp orine sildena fil Swarfari n erythro mycin itracon azole hydrox yitracon azole ritonav ir 200 q.d. formed from itracona zole diltiaze m desmet hyldilti azem verapa mil clarith romyci n saquin avir phenyt oin carba mazepi ne 500 b.i.d. 60 t.i.d. formed from diltiaze m 120 t.i.d. 250 b.i.d. 1200 t.i.d. 300 q.d. 400 b.i.d. carba mazepi ne10,11epoxid e formed from carbam azepine 400 q.d. or 200 b.i.d. 600 q.d. 100 q.d. 20 t.i.d. 25 q.d. 500 t.i.d. 531.4 823 1202 474.6 308.3 733.9 705.6 721.7 720.95 414.5 400 454.6 748 670.86 252.28 236.27 252.27 3.28 4.3 2.97 2.7 2.5 5.66 4.78 4.3 2.8 2.01 3.81 1.7 3.9 2.47 2.22 1.44 7.9 - 6.5 - 8.8 3.7 2.5, 4.9 2.0 8.1 8.06 8.9 8.99 6.67 - - - acidic pKa 4.04 2.94, 6.51 - 1.7 - - 5.1 - - - - - - - - 10.94 8.15 - - fu,p 0.029 0.15 0.037 0.036 0.009 0.31 0.016 0.021 0.02 0.25 0.323 0.10 0.18 0.025 0.1 0.25 0.48 blood to plasma ratio 0.62 0.90 1.36 0.63 0.59 0.85 0.58 1 0.59 0.96 1 0.76 1 0.6 0.61 1.1 1.53 1 NA 1 1 1 0.9 0.84 NA oral dose (mg) and dosing frequency molecular weight (g/mol) lipophilicity (logP) basic pKa fa 1 1 1 1 1 1 1 NA 1 (h-1) 0.78 0.51 1.49 2.58 1.85 0.52 0.62 NA 0.24 6 NA 1.22 2.37 2.55 0.53 0.5 NA Qgut (L/h) 13.2 10 8.16 10 11.6 1.67 15.7 NA 11.2 13.5 NA 14.4 9.34 4.91 13.2 12.6 NA fu,gut 0.06 0.15 1 0.036 1 1 0.016 1 0.02 0.25 1 1 1 0.025 1 1 1 0.345 0.015 / 0.97 0.33 5.6 3.46 0.114 0.75 10.7 69.5 3.09 3.9 3.4 1.75 0.78 0.78 1.5 - - 82 / 0.9 0.0013 0.0144 36.1 2.43 - 2.43 3.63 3.5 / 0.58 0.57 10.5 0.41 0.025 / 0.662 - - - - - - - - 23.2 - - - 4.75 1.74 2.21 12 - - - - - - - - - 2.25 - - - 0.70 1.09 2 2.13 - - - - - 0.32 - - - - - - - - - - - - 7.7 - 8 - - - - - - - - - - - - 6.2 slope 0.16 / h slope 0.14 / h [8, 9] [10] [11] [12, 13] [18] [14] [14] [19] ka Vss (L/kg) Ki CYP3A4 (µM) / fu,inc KI CYP3A4 (µM) / fu,inc kinact CYP3A4 (h-1) IndC50 CYP3A4 (µM) Indmax CYP3A4 (fold) reference* [14] [15] [14, 16] [17] [10] * Describing the details such as data source and/or performance of the PBPK models; data are from unchanged Simcyp v12 compound library PBPK model files, with the exception of the fu,gut of sildenafil, which was set equal to its plasma protein binding and the CYP3A4 Ki for cyclosporine [20]. Absorption was modeled using the 1st order model. Distribution was modeled using a minimal PBPK model. Free fraction of the in vitro input data was 1, unless indicated otherwise. fu,p, fraction of unbound drug in plasma; pKa, acid dissociation constant; fa, fraction absorbed; Qgut hybrid parameter of enterocytic blood flow and drug permeability; f u,gut, free fraction in the enterocyte; Vss apparent volume of distribution at steady state; K i inhibition constant; fu,inc, free fraction in the incubation; KI, concentration at half-maximal kinact; kinact maximum rate constant for inactivation; IndC50, inducer concentration at half-maximal Indmax; Indmax, maximal induction; NA, not applicable. 6 Summary of the used interacting drug PBPK models - elimination drug ketoconazole rifampicin cyclosporine sildenafil S-warfarin erythromycin itraconazole hydroxyitraconazole ritonavir diltiazem desmethyldiltiazem verapamil clarithromycin saquinavir phenytoin carbamazepine carbamazepine10,11-epoxide elimination mechanism active uptake into hepatocytes was 2.07 fold and oral clearance of 7.4 L/h and a renal clearance of 0.147 L/h. i.v. clearance of 7 L/h and a renal clearance of 1.2 L/h. CYP3A4 with a Km of 1.17 μM and a Vmax of 200 pmol/min/pmol enzyme and a renal clearance of 0.018 L/h. CYP2C9 and CYP3A4 using a CLint 0.486 and 2.2 μL/min/pmol enzyme, respectively and a renal clearance of 0.72 L/h. renal and polymorphic specific CYP2C9 clearance as described previously. i.v. CL of 27.8 L/h and a renal clearance of 3.13 L/h. CYP3A4 with a Km of 0.039 μM and a Vmax of 0.065 pmol/min/pmol enzyme towards its metabolite hydroxyitraconazole. CYP3A4 with a Km of 0.027 μM and a Vmax of 0.13 pmol/min/pmol enzyme. oral clearance of 9.1 L/h and a renal clearance of 0.32 L/h. CYP3A4 with a Km of 50 μM and a Vmax of 1880 pmol/min/pmol enzyme and by CYP1A2 using a CLint 2.4 μL/min/pmol enzyme towards desmethyldiltiazem and using a human liver microsomal CLint of 109 μL/min/mg protein. CYP3A4 using a CLint 43.2 μL/min/pmol, using a human liver microsomal CLint of 1.65 μL/min/mg protein and a renal clearance of 2.88 L/h. i.v. CL of 52.2 L/h and a renal clearance of 2.37 L/h. CYP3A4 using two pathways, with a K m of 20.6 and 63.9 μM and a V max of 37.9 and 63.9 pmol/min/pmol enzyme, respectively and a renal clearance of 8.05 L/h. CYP3A4 with a Km of 0.51 μM and a Vmax of 2860 pmol/min/pmol enzyme and a renal clearance of 0.95 L/h. CYP2C9 and CYP2C19 with a Km of 4.1 and 1.53 μM and a Vmax of 0.24 and 1.53 pmol/min/pmol enzyme, respectively, using a human liver microsomal CLint of 0.97 μL/min/mg protein and a renal clearance of 0.015 L/h. CYP3A4, CYP3A5 and CYP2C8 with a K m of 180, 332 and 741 μM and a Vmax of 0.72, 1.44 and 0.03 pmol/min/pmol enzyme, respectively, towards its metabolite carbamazepine-10,11-epoxide and additional clearance by CYP3A4 using a CLint of 0.0106 µL/min/pmol enzyme, a human liver microsomal CLint of 0.255 μL/min/mg protein and a renal clearance of 0.0084 L/h. reference* [21] [10] [11] [12, 13] [22] [14] [15] [16] [17] [18] [17] [16] [19] [23] oral clearance of 6.07 L/h and a renal clearance of 0.14 L/h. * Describing the details of the compound PBPK model elimination processes; data are from unchanged Simcyp v12 compound library PBPK model files i.v. intravenous, Km Michaelis-Menten constant, Vmax maximum rate CLint intrinsic clearance of drug from plasma References cited in the supplementary material 1. OECD. Guidelines for the Testing of Chemicals. Test No. 107: Partition Coefficient (n-octanol/water): Shake Flask Method; 1995. 2. Allen R, Box K, Comer J, Peake C, Tam K. Multiwavelength spectrophotometric determination of acid dissociation constants of ionizable drugs. Journal of pharmaceutical and biomedical analysis. 1998;17:699-712. 3. Riedel J. Distribution - in vitro tests - protein binding. In: Riedel J, editor. Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays: Springer Science & Business Media; 2006. p. 480-1. 4. Bruderer S, Aanismaa P, Homery MC, Hausler S, Landskroner K, Sidharta PN, et al. Effect of cyclosporine and rifampin on the pharmacokinetics of macitentan, a tissue-targeting dual endothelin receptor antagonist. The AAPS journal. 2012;14:68-78. 5. Atsmon J, Dingemanse J, Shaikevich D, Volokhov I, Sidharta PN. Investigation of the effects of ketoconazole on the pharmacokinetics of macitentan, a novel dual endothelin receptor antagonist, in healthy subjects. Clinical pharmacokinetics. 2013;52:685-92. 6. Sidharta PN, van Giersbergen PL, Wolzt M, Dingemanse J. Investigation of mutual pharmacokinetic interactions between macitentan, a novel endothelin receptor antagonist, and sildenafil in healthy subjects. British journal of clinical pharmacology. 2014;78:1035-42. 7. Sidharta PN, Dietrich H, Dingemanse J. Investigation of the effect of macitentan on the pharmacokinetics and pharmacodynamics of warfarin in healthy male subjects. Clinical drug investigation. 2014;34:545-52. 8. Perdaems N, Blasco H, Vinson C, Chenel M, Whalley S, Cazade F, et al. Predictions of metabolic drug-drug interactions using physiologically based modelling. Clinical pharmacokinetics. 2010;49:239-58. 9. Rowland Yeo K, Walsky RL, Jamei M, Rostami-Hodjegan A, Tucker GT. Prediction of time-dependent CYP3A4 drug-drug interactions by physiologically based pharmacokinetic modelling: impact of inactivation parameters and enzyme turnover. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences. 2011;43:160-73. 10. Xu Y, Zhou Y, Hayashi M, Shou M, Skiles GL. Simulation of clinical drug-drug interactions from hepatocyte CYP3A4 induction data and its potential utility in trial designs. Drug Metabolism and Disposition. 2011;39:1139-48. 11. Jamei M, Bajot F, Neuhoff S, Barter Z, Yang J, Rostami-Hodjegan A, et al. A mechanistic framework for in vitro–in vivo extrapolation of liver membrane transporters: prediction of drug–drug interaction between rosuvastatin and cyclosporine. Clinical pharmacokinetics. 2014;53:73-87. 12. Zhao P, Vieira MdL, Grillo JA, Song P, Wu TC, Zheng JH, et al. Evaluation of exposure change of nonrenally eliminated drugs in patients with chronic kidney disease using physiologically based pharmacokinetic modeling and simulation. The Journal of Clinical Pharmacology. 2012;52:91S-108S. 13. Hsien L. Identifying paediatric needs in cardiology and the prediction of sildenafil exposure in children with pulmonary arterial hypertension: Heinrich-Heine-Universität Düsseldorf; 2010. 14. Wang Y-H. Confidence assessment of the Simcyp time-based approach and a static mathematical model in predicting clinical drug-drug interactions for mechanism-based CYP3A inhibitors. Drug Metabolism and Disposition. 2010;38:1094-104. 8 15. Guest EJ, Rowland‐Yeo K, Rostami‐Hodjegan A, Tucker GT, Houston JB, Galetin A. Assessment of algorithms for predicting drug–drug interactions via inhibition mechanisms: comparison of dynamic and static models. British journal of clinical pharmacology. 2011;71:72-87. 16. Almond L, Yeo KR, Howgate E, Tucker GT, Rostami-Hodjegan A. Mechanistic prediction of HIV drug-drug interactions in virtual populations from in vitro enzyme kinetic data: ritonavir and saquinavir. International Workshop on Clinical Pharmacology of HIV Therapy, Budapest, Hungary, 16th–18th April; 2007; 2007. 17. Yeo KR, Jamei M, Yang J, Tucker GT, Rostami-Hodjegan A. Physiologically based mechanistic modelling to predict complex drug–drug interactions involving simultaneous competitive and time-dependent enzyme inhibition by parent compound and its metabolite in both liver and gut—the effect of diltiazem on the time-course of exposure to triazolam. European Journal of Pharmaceutical Sciences. 2010;39:298-309. 18. Neuhoff S, Yeo KR, Barter Z, Jamei M, Turner DB, Rostami‐Hodjegan A. Application of permeability‐limited physiologically‐based pharmacokinetic models: Part II‐ prediction of p‐glycoprotein mediated drug–drug interactions with digoxin. Journal of pharmaceutical sciences. 2013;102:3161-73. 19. Polasek TM, Polak S, Doogue MP, Rostami-Hodjegan A, Miners JO. Assessment of inter-individual variability in predicted phenytoin clearance. European journal of clinical pharmacology. 2009;65:1203-10. 20. Gertz M, Cartwright CM, Hobbs MJ, Kenworthy KE, Rowland M, Houston JB, et al. Cyclosporine inhibition of hepatic and intestinal CYP3A4, uptake and efflux transporters: application of PBPK modeling in the assessment of drug-drug interaction potential. Pharmaceutical research. 2013;30:761-80. 21. Huang Y, Colaizzi J, Bierman R, Woestenborghs R, Heykants J. Pharmacokinetics and dose proportionality of ketoconazole in normal volunteers. Antimicrobial agents and chemotherapy. 1986;30:206-10. 22. Almond LM, Rowland-Yeo K, Howgate EM, Tucker GT, Rostami-Hodjegan A. Prediction of the oral clearance of S-warfarin in CYP2C9 genotypes from in vitro enzyme kinetic data. 9th European ISSX Meeting, Manchester, UK, 4th–7th June In: Drug Metab Rev; 2006; 2006. p. S92-3. 23. Vajjah P, Jamei M, Johnson T, Neuhoff S, Rostami-Hodjegan A. Prediction of serum and cerebrospinal fluid concentrations of carbamazepine: An application of parameter estimation and the permeability limited 4-compartment brain model in Simcyp®. 21st PAGE Meeting. Venice, Italy. 9