Web Resources
advertisement

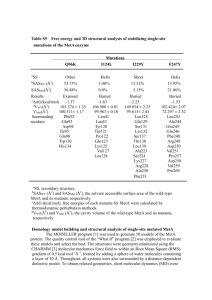
1 A TFG mutation causes autosomal dominant axonal Charcot-Marie-Tooth disease Pei-Chien Tsai,1,2,3,13 Yen-Hua Huang,4,5,13 Yuh-Cherng Guo,6,7 Hung-Ta Wu,8 Kon-Ping Lin,1,2 Yi-Chu Liao,1,2 Cheng-Tsung Hsiao,1,2 Yi-Chun Lu,1,2 Tze-Tze Liu,9 Shaw-Fang Yet,10 Ming-Ji Fann,3,11 Lun-Sen Kao,3,9,11 Bing-Wen Soong,1,2,3,12,* Yi-Chung Lee1,2,3,* 1 Department of Neurology, Taipei Veterans General Hospital, Taipei 11217, Taiwan; 2 Department of Neurology, National Yang-Ming University School of Medicine, Taipei 11221, Taiwan; 3 Brain Research Center, National Yang-Ming University, Taipei 11221, Taiwan; 4Department of Biochemistry, National Yang-Ming University School of Medicine, Taipei 11221, Taiwan; 5Center for Systems and Synthetic Biology, National Yang-Ming University, Taipei 11221, Taiwan; 6Department of Neurology, China Medical University Hospital, Taichung 40447, Taiwan; 7Instute of Clinical Medicine, National Yang-Ming University School of Medicine, Taipei 11221, Taiwan; 8Department of Radiology, Taipei Veterans General Hospital, Taipei 11217, Taiwan; 9Department of Life Sciences and Institute of Genome Sciences, National Yang-Ming University, Taipei 11221, Taiwan; 10Institute of Cellular and System Medicine, National Health Research Institues, Zhunan 35053, Taiwan; 11Genome Research Center, National Yang-Ming University, Taipei 11221, Taiwan;12Institute of Neuroscience, National Yang-Ming University, Taipei 11221, Taiwan; 13These authors contributed equally to this work. *Correspondence: ycli@vghtpe.gov.tw (Y.-C.L.) and bwsoong@ym.edu.tw (B.-W.S.) 2 Words in the main body: 3520 Abstract words count: 219 Short title: A TFG mutation causes AD-CMT2 3 Abstract Charcot-Marie-Tooth disease (CMT) is a group of inherited neuropathies sharing common features of progressive distal muscle atrophy and weakness, sensory loss, foot deformities and depressed tendon reflexes. Mutations in approximately 60 genes have been identified as being associated with CMT. Nevertheless, the genetic etiologies of at least 30% of CMTs have yet to be elucidated. We studied a five-generation pedigree with the autosomal dominant axonal CMT (CMT2) and excluded mutations in common CMT2 implicated-genes by Sanger sequencing. Subsequent exome sequencing of two affected individuals revealed a novel heterozygous mutation, c.806G>T (p.Gly269Val), in the TRK-fused gene (TFG) that perfectly co-segregates with the CMT2 phenotype in all 27 family members. This mutation occurs at an evolutionarily conserved residue and is absent in the 1,140 ethnically matched control chromosomes. Cell transfection studies showed that the TFG p.Gly269Val mutation increased the propensity of TFG proteins to form aggregates, which sequestrated both mutant and wild-type TFG and might deplete functional TFG molecules. The secreted Gaussia luciferase reporter assay demonstrated that inhibition of endogenous TFG resulting in compromised protein secretion pathways, which could be rescued by expressing wild-type TFG but not the p.Gly269Val altered proteins. This study identifies a novel cause of CMT2 and highlights the importance of TFG in protein secretory pathways which are essential for proper functioning of human peripheral nervous system. Keywords: Charcot-Marie-Tooth disease, CMT, exome sequencing, TFG, TRK-fused gene, CMT2. 4 Charcot-Marie-Tooth disease (CMT), also called Hereditary Motor and Sensory Neuropathy (HMSN), is a clinically and genetically heterogeneous group of inherited neuropathy sharing common features of progressive distal muscle atrophy and weakness, distal sensory loss, foot deformities and depressed tendon reflexes. It is one of the most common inherited neurological disorders with a prevalence of about 1/2500. CMT can be classified into a demyelinating or axonal subtype according to its electrophysiological or pathological features. Usually, in nerve conduction studies, demyelinating CMT (CMT1) is associated with significantly reduced nerve conduction velocities (NCVs), whereas axonal CMT (CMT2) is accompanied with reduced amplitudes and relatively preserved NCVs.1-3 Clinically, median motor NCV is the most commonly used parameter, with a cut-off value of 38 m/s to separate CMT1 from CMT2.4 Mutations in more than 60 genes have been identified in CMT to date and at least 15 of them are known for typical dominant CMT2, but the genetic causes of more than 60% of CMT2 remain elusive.1-3 We studied a large Taiwanese family of Han Chinese origin and enrolled 27 members, including 16 individuals affected with autosomal-dominant CMT2, nine unaffected individuals, and two married-in spouses (Figure 1A). Disease status was ascertained by neurological examination and electrophysiological evaluation. The age at disease onset ranged from 28 to 40 years. All affected individuals presented with typical CMT2 phenotypes, including slowly progressive symmetrical muscle atrophy and weakness from distal parts of limbs and mild distal sensory loss. The clinical severity varied from being asymptomatic to being dependent on the assistance of a walker for ambulation. The median motor NCVs ranged from 43.6 to 60.8 m/s. Clinical manifestations of part of the affected individuals are shown in Table 1. Written informed consents were obtained from all subjects and the study was 5 approved by the Institutional Review Board of Taipei Veterans General Hospital, Taiwan. Genomic DNA was isolated from peripheral blood leukocytes following a standard protocol. The clinical features of the proband (IV-2) are used to examplify the disease in this family. IV-2 is a 36-year-old man who had manifested slowly progressive weakness of distal lower limbs since the age of 28 years. He started to walk at a normal age. Physical examination at age 36 revealed high-arched feet, hammer toes, generalized areflexia, atrophy and weakness of the intrinsic muscles of the hands (score 4/5 in Medical Research Council Scale), feet (2/5) and also the muscles in the legs, resulting in impaired dorsiflexion and plantar flexion of the feet (4 and 4+/5). Despite a lack of sensory complaints, mildly diminished sensation for all modalities in regions distal to the ankles was observed. Nerve conduction studies demonstrated the presence of an axonal sensorimotor polyneuropathy with normal NCVs but reduced amplitudes. A sural nerve biopsy revealed loss of large myelinated nerve fibers (MFs) with relatively preserved median and small MFs (fiber density: 4366/mm2; fibers with diameters larger than 7 um: 23.7%) and no signs of demyelination (Figure 1B and 1C). Sanger sequencing excluded mutations in the majority of the genes known to be putatively involved in CMT2;5 MFN2 (MIM 608507), RAB7 (MIM 602298), TRPV4 (MIM 605427), GARS (MIM 600287), NEFL (MIM 162280), HSPB1 (MIM 602195), MPZ (MIM 159440), GDAP1 (MIM 606598), HSPB8 (MIM 608014), DNM2 (MIM 602378), and AARS (MIM 601065). To identify the genetic cause of the CMT2 in this family, we employed a whole-exome sequencing strategy as previously described.6 In brief, exonic regions from the genomic DNA of the two affected individuals of the family (III-8 and IV-3 in Figure 1A) were captured and enriched using the Agilent SureSelect Human All Exon 6 v2 kit. Then, enriched DNA was sequenced on an Illumina Genome Analyzer GAII with 100 bp paired-end reads after standard sample preparation. Overall, an average of 30.8 Gb sequence was generated per individual, resulting in an average coverage depth of 398.1x per targeted base. After analysis and filtering (Table S1), 109 heterozygous coding variants were found to be shared by both patients and were not present in the dbSNP, the 1000 Genomes Project,7 or the in-house exome data from 10 non-CMT ethnic controls. Sanger sequencing of these variants in other 25 members of the family revealed that only one variant, c.806G>T (p.Gly269Val) in the TRK-fused gene (TFG; MIM 602498) (RefSeq accession number NM_006070.5), perfectly segregated with the CMT phenotype (Figure 1A and 1D). The pathogenic role of the mutation, TFG p.Gly269Val, is further supported by its absence in the 1,140 ethnically matched control chromosomes and 6500 exomes in the Exome Variant Server of the NHLBI Exome Sequencing Project, involvement of a highly conserved residue (Figure S1) and being pathogenic as predicted by four different programs: SNPs&GO,8 Panther,9 PMut10 and SNAP.11 Mutational analyses of TFG were further conducted in 21 unrelated individuals with molecularly unassigned CMT2 and no new family was found. Since the CMT2 individuals in this study were selected from a continuous series of 251 CMT families after an extensive mutational analysis and electrophysiological evaluation for CMT,5 the frequency of TFG mutation in our CMT population was approximately 0.4% (1/251). TFG was initially identified as a part of the neurotrophic tyrosine kinase receptor type 1 (NTRK1)-derived thyroid oncogene, TRK-T3, whose protein product is a chimera consisting of the N-terminal portion of the protein TFG and the C-terminus of the tyrosine kinase NTRK1.12 Subsequent studies found additional TFG fusion oncogenes in other human malignancies.13,14 TFG is ubiquitously expressed in normal 7 human tissues and localizes to endoplasmic reticulum (ER) exit sites in an oligomeric form.15,16 It interacts with Sec16B and has an important role in the COPII-mediated export of secretory cargo from the ER.16 Inhibition of TFG in cell lines compromises the protein secretion from ER and alters the ER structure.17 Two mutations in TFG were recently implicated in two neurological diseases, including a homozygous p.Arg106Cys mutation in complicated hereditary spastic paraplegia and,17 interestingly, a heterozygous p.Pro285Leu mutation in hereditary motor and sensory neuropathy with proximal dominant involvement (HMSN-P; MIM 60448).15 HMSN-P is an adult-onset autosomal dominant neuropathy characterized by proximal predominant muscle weakness and atrophy, widespread fasciculations, muscle cramps, and late onset of distal sensory involvement. In contrast to the typical clinical manifestations of CMT presenting with predominantly distal muscle atrophy and weakness, the clinical presentations of HMSN-P are more similar to those of amyotrophic lateral sclerosis.15 The TFG p.Pro285Leu mutation has been reported in four Japanese families and one Korean family with HMSN-P.15,18 To affirm that the phenotype of the affected individuals harboring the TFG p.Gly269Val mutation in this family is typical CMT rather than HMSN-P, we utilized skeletal muscle magnetic resonance imaging (MRI) to scrutinize the distribution of their lower limb muscle atrophy. Two affected individuals (IV-2, IV-16) and one healthy control subject were studied with MRI of the hip and lower limbs by using a 1.5-T MR unit (Signa CV/I, GE Medical Systems, Milwaukee, WI). T1-weighted images [517-613 ms repetition time (TR), 7.98-8.34 ms echo time (TE), 10 mm thickness] were used to look for the presence of muscle atrophy with fatty substitution. Both affected individuals had a distally predominant muscle atrophy and fat replacement with a proximal-to-distal gradient of muscle involvement on 8 T1-weighted MRIs (Figure 2), compatible with the features of typical CMT. Individual IV-16, who was only mildly affected, had a mild fatty infiltration of muscles in the legs (mainly gastrocnemius and soleus muscles) and a minimal involvement of muscles in the thighs (mainly vastus lateralis muscle) (Figure 2B). Individual IV-2, who was markedly affected, had a severe and widespread muscle atrophy with fat replacement in the legs (most severe in gastrocnemius and soleus), a moderate degree of muscle involvement in the thighs (mainly vasti, biceps femoris, and semitendinosus muscles), and a mild fatty infiltration of gluteal maximus muscle (Figure 2C). To explore the influence of the p.Gly269Val mutation on the TFG function, we conducted in vitro transfection analysis. We subcloned a 1.2 kb PCR-amplified fragment containing the coding region of TFG from human cDNA libraries into pFLAG-CMV-5a (Sigma-Aldrich) to construct TFG expression plasmids (Table S2). The mutation c.806G>T (p.Gly269Val) was introduced by using QuikChange Site-Directed Mutagenesis method (Stratagene) (Table S2). To investigate the expression of both wild-type and mutant TFG proteins, we incubated human embryonic kidney (HEK) 293 cells maintained in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone) containing 10% fetal bovine serum (FBS; Invitrogen) at 37oC and in 5% CO2 and transfected them with plasmids expressing wild-type or Gly269Val TFG by using calcium-phosphate precipitation. At 48 hours after transfection, total-protein cell extracts were analyzed by Western blotting analysis, which showed that the steady-state level of the Gly269Val mutant TFG was significantly lower than that of wild-type TFG (Figure 3A, 3B; Table S3). Since reduced steady-state level of mutant TFG proteins may result from an enhanced protein degradation, a reduced mRNA synthesis, or formation of detergent 9 insoluble protein aggregates19, we next evaluated the impact of the p.Gly269Val mutation on TFG degradation by a cycloheximide-chase assay as previously described,6 and found that the mutation did not compromise the protein stability (Figure S2). Further analysis of TFG mRNA levels in HEK293 cells expressing wild-type or Gly269Val TFG by real-time quantitative PCR analysis (RT-qPCR) revealed no significant difference between the wild-type and the mutant TFG mRNA levels (Figure 3C; Table S2). The TFG mRNA levels of the dermal fibroblasts from the affected individual IV-2 and an age-matched male healthy control subject were also similar (Figure S3), indicating that the p.Gly269Val mutation does not affect the quantity of TFG mRNA. Then, we utilized Aggrescan, a bioinformatic tool, to evaluate whether the mutation may increase the propensity of TFG proteins to form aggregates.20 In silico analysis predicted that the TFG p.Gly269Val mutation might increase the aggregation tendency of TFG by generating a new aggregation-prone region at the residues 269 to 274. To elucidate whether the reduced levels of Gly269Val TFG in the transfected cells is resulting from the enhanced tendency of the TFG mutant proteins to form aggregates, we examined the solubility of wild-type TFG and TFG mutants in HEK293 cells. We also used Pro285Leu TFG as a control because previous study showed that its expression level in HEK293T cells was not different from that of wild-type TFG.18 The cells transfected with different TFG constructs were extracted with RIPA buffer (soluble fraction) first and subsequently extracted with urea buffer (7M urea, 2M thiourea, 1% ASB-14, 40mM Tris, pH 8.5) to recover insoluble TFG. Western blotting analysis revealed that only the Gly269Val TFG was predominantly found in the insoluble fraction of cell lysates, whereas the wild-type protein and the Pro285Leu mutant were detected predominantly in the soluble fraction (Figure 4A 10 and 4B). Similar results were obtained when experiments were performed using HeLa cells (Figure 4A and 4B), indicating that the Gly269Val TFG had such a strong tendency to form detergent-insoluble aggregates that its soluble form in cytosol was significantly depleted, while the Pro285Leu TFG as well as wild-type TFG did not have such propensities. The differences in biochemical characteristics between Gly269Val TFG and Pro285Leu TFG may partially explain why they are associated with disparate clinical phenotypes (CMT2 and HMSN-P). To clarify the intracellular features of both wild-type and Gly269Val TFG, we performed immunofluorescence analyses of HEK293 cells transfected with wild-type or the mutant TFG constructs (Table S3). Cell expressing wild-type TFG displayed numerous punctate TFG-specific staining throughout the cytosol, whereas cells expressing the Gly269Val TFG demonstrated multiple large cytoplasmic aggregates with TFG-specific staining (Figure 3D). These cytoplasmic aggregates were negative for TDP-43 staining (data not shown). Both wild-type TFG and Gly269Val TFG had a similar subcellular localization and were mainly expressed in the ER, evidenced by both colocalizing with DesRed-ER (Clontech), an ER marker (Figure S4A), and Sec16B, a marker of ER exit sites (Figure S4B).16 Since wild-type TFG molecules form oligomers in their normal functioning, we investigated the association between wild-type TFG and Gly269Val TFG proteins. Equal amounts of plasmids encoding DesRed-tagged wild-type TFG and GFP-tagged Gly269Val TFG were co-transfected into HEK293 cells. Co-localization of wild-type and mutant TFG within cytoplasmic aggregates suggests that the Gly269Val TFG molecules did not lose their ability to congregate with wild-type TFG in an oligomeric complex (Figure S4C). Since the fluorescent analysis revealed that wild-type TFG and the Gly269Val mutant co-localized within the cytoplasmic aggregates in the cells (Figure S4C), the 11 mutant TFG may incorporate the wild-type protein to form aggregates. To evaluate the impact of such effect on cells harboring the heterozygous p.Gly269Val TFG mutation, we compared the level of soluble cytoplasmic TFG of HEK293 cells transfected with wild-type TFG construct and that of cells transfected with half dose of the wild-type construct and half dose of the mutant construct. Western blotting analysis revealed that the TFG level of the cells expressing both wild-type and mutant TFG was only approximately one third of that of the cells expressing solely wild-type TFG (Figure S5A and B), indicating that the Gly269Val mutant may partially deplete wild-type TFG through the aggregate formation process. In addition, accumulated cytoplasmic protein aggregates may provoke ER stress or lead to cellular toxicity. Thus, we examined whether the Gly269Val TFG mutants affected cell viability. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay was used to assess cell viability of HEK293 cells transfected with vectors expressing wild-type TFG or Gly269Val TFG, or empty vector. We found that cells expressing Gly269Val TFG did not have significant reduction in viability when compared with the cells expressing wild-type TFG or the cells transfected with empty vector (Figure S6A), suggesting that Gly269Val TFG did not have a significant cellular toxicity. To investigate whether the Gly269Val mutant may induce ER stress or activate the unfolded protein response (UPR), biomarkers of ER stress, such as expression of ER resident chaperon Bing Ig protein (BiP)21 and transcriptional factor CAATT enhancer-binding protein homologous protein (CHOP)22 and alternative splicing of X-box binding protein I (XBP-1),23 were examined in the transfected HEK293 cells (Table S2). Cells exposed to a stress stimulator dithiothreitol (DTT) or transfected to express Arg89Cys myelin protein zero, a previously identified ER stress-inducing 12 mutant protein,24 were used as the positive controls. As shown in Figure S6B, cells expressing Gly269Val TFG did not have a significantly higher level of Bip or spliced XBP-1 compared with the cells expressing wild-type TFG, suggesting that the Gly269Val TFG did not induce ER stress, or increase the cellular susceptibility to ER stress. Under treatment of 1 mM DTT, the levels of BiP, CHOP, or spliced XBP-1 did not increase in the cells expressing Gly269TFG compared with those expressing wild-type TFG (Figure S6C). Because Gly269Val TFG may partially deplete wild-type TFG and its overexpression did not increase cellular toxicity, the pathogenesis of the heterozygous TFG p.Gly269Val mutation may come from low soluble level of cytoplasmic TFG, which may result in insufficient functional TFG molecules. To test the impact of depletion of wild-type TFG on cells, we designed siRNA specifically targeting the 3’UTR of TFG to efficiently suppress the expression of endogenous wild-type TFG (Figure S7, Table S2). Utilizing MTT assay, we examined the cell viability of the HEK293 cells co-transfected with the siRNA and plasmids expressing wild-type TFG or Gly269Val TFG, or empty vectors, and found that depletion of endogenous TFG significantly reduced in vitro viability, which could be rescued by expressing exogenous wild-type TFG but not Gly269Val TFG (Figure S8). Previous studies have shown that TFG depletion slows protein secretion from the ER and alters the ER structure.16,17 To investigate the effect of the Gly269Val mutant on the protein secretory pathway, we co-transfected HEK293 cells with reporter plasmids encoding Gaussia luciferase (Gluc) and Firefly luciferase (Fluc), along with plasmids expressing wild-type or Gly269Val TFG or empty vector. The Gaussia luciferase can be secreted out of the cells and has been commonly utilized to monitor protein secretory pathway25. We measured the Gluc activities in the culture medium 13 of the cells at 48 hours after transfection with wild-type, Gly269Val TFG, or empty constructs, and normalized the Gluc activities with the corresponding internal Fluc control activities. The luciferase assay revealed that there was no significant difference between the secretory efficiencies of Gluc between the cells expressing wild-type TFG, mutant TFG, or empty vector (Figure 5A). Since the endogenous TFG of the HEK293 cells might have been sufficient for the protein secretory function, the siRNA targeting the 3’UTR of TFG was used to inhibit endogenous TFG of cells transfected with different TFG constructs or empty vectors and the secretion efficiencies of Gluc in cells were investigated. The results indicated that inhibition of endogenous TFG compromised the protein secretory pathway, which could be rescued by replenishing with exogenous wild-type TFG but not Gly269Val TFG (Figure 5B). These findings support that cells with a heterozygous TFG p.Gly269Val mutation may have compromised protein secretory function, which may lead to a decrease in cellular viability. Utilizing exome sequencing, bioinformatics filtering and co-segreation analysis, we have identified only one heterozygous mutation, TFG p.Gly269Val, in this large pedigree with autosomal dominant CMT2. This mutation alters the conserved amino acid residue, is absent in the 6,500 exomes from the NHLBI Exome Variant Server, and does not occur in 1,140 ethnic control chromosomes. In vitro transfection studies revealed that Gly269Val TFG mutant proteins tended to form large intracellular aggregates and depleted functional TFG molecules by sequestrating wild-type TFG into aggregates. Depletion of TFG in cells by siRNA compromised the protein secretory function and decreased cell viability, which both could be rescued by expression of exogenous wild-type TFG but not the Gly269Val mutant. This study provides strong genetic and functional evidences to support that the TFG p.Gly269Val 14 mutation causes CMT2 and thus, identifies TFG as a new member on the list of CMT2-associated genes. TFG is ubiquitously expressed in human neuronal and non-neuronal cells, as well as in the spinal motor neurons and dorsal root ganglia.15 Only two mutations in TFG have been previously identified. One is the heterozygous p.Pro285Leu mutation which causes HMSN-P and another is the homozygous p.Arg106Cys mutation which results in a complicated form of recessive HSP, presenting as spastic paraplegia, optic atrophy, and neuropathy (SPOAN).15,17 Both mutations are associated with neurological diseases, indicating that TFG is critically important for neuronal function. Our work provides additional genetic evidence that the heterozygous p.Gly269Val mutation in TFG could cause a dominant axonal CMT. Thus far, more than 60% of the genetic causes of axonal CMT remain unknown, reflecting the molecular complexity of the structure and function of peripheral axons and neurons. Although the full scopes of TFG functions are still unclear, recent studies revealed that TFG functions at ER exit sites (ERES) and regulates secretory protein vesicle biogenesis and egress from the ER.16 In neuronal cells, properly functioning ERES helps provide sufficient protein cargoes for the axonal transport system and enable efficient secretion of locally synthesized proteins from the axonal ER.26 ERES also plays an important role in supporting axonal outgrowth.27 Therefore, TFG dysfunction may compromise protein secretory pathway and result in a shortage or insufficiency of local functional proteins in axons, which may be the common consequence of a host of hereditary axonopathies associated with mutations in the genes involved in axonal transport (such as HSPB1, NEFL, BICD2, DYNC1H1, KIF5A, KIF1A, MYH14), or ER and Golgi organization (such as BSCL2, REEP1, ATL1, FAM134B).3,28 It is intriguing that three different mutations in TFG cause three disparate clinical 15 phenotypes, implying that each mutation has its unique pathogenic mechanisms. The TFG p.Gly269Val mutation, which is associated with a dominant CMT2, increases the aggregation propensity of TFG proteins. In cells harboring the heterozygous p.Gly269Val mutation, mutant TFG proteins tend to form cytoplasmic aggregates, which also sequestrate a sizable wild-type TFG proteins and lead to insufficient soluble TFG molecules available to maintain their proper functions. The TFG p.Gly269Val mutation seems not to have a toxic gain-of-function, demonstrated by that in vitro overexpression of the mutant TFG did not affect cell viability. However, its dominant-negative effect in neuronal cells with the heterozygous mutation may deplete wild-type TFG molecules and compromise protein secretory process, with the neurons with the longest axons most vulnerable and leading to the distal symmetric motor and sensory symptoms in the CMT2. The TFG p.Arg106Cys mutation, which is associated with a recessive complicated HSP presenting as SPOAN, abolishes the ability of TFG proteins to self-assemble into a functional oligomer.17 Individuals carrying heterozygous TFG p.Arg106Cys mutation are asymptomatic, suggesting that one copy of wild-type TFG is sufficient to produce enough proteins for physiological function. Compared with those harboring the heterozygous TFG p.Gly269Val mutation, the individuals carrying the homozygous TFG p.Arg106Cys mutation may have a much more profound loss of TFG function, which thus causes a widespread axonopathy involving corticospinal tracts, peripheral nerves and optic nerves. In addition, the heterozygous TFG p.Pro285Leu mutation involved in dominant HMSN-P may pose a toxic gain-of-function effect through a possible RNA-mediated and TDP-43 related mechanism.15 The Pro285Leu TFG protein may elicit a direct toxic effect on neuronal cells to cause the motor and sensory symptoms. The explanation for selective proximal involvement in HSMN-P remains elusive. 16 In conclusion, our findings demonstrate a novel TFG mutation causing autosomal dominant CMT2 and highlight the importance of TFG in protein secretory pathways which are essential for proper functioning of human peripheral nervous system. Supplemental Data Supplemental Data, including eight figures and three tables, can be found with this article online at http://www.cell.com/AJHG/. Acknowledgments The authors thank the patients who participated in this study. This work was supported by research grants from the National Science Council, Taiwan, ROC (NSC102-2628-B-075-006-MY3), the Ministry of Education, Aim for the Top University Plan (V100-E6-006, V100-E6-007 and V101E7-005), the Brain Research Center, National Yang-Ming University, the High-throughput Genome Analysis Core Facility and Bioinformatics Consortium of Taiwan of National Core Facility Program for Biotechnology, Taiwan (NSC100-2319-B-010-001 NSC100-2319-B-010-002). Web Resources The URLs for data presented herein are as follows. 1000 Genomes Project, http://www.1000genomes.org Aggrescan, http://bioinf.uab.es/aap/ dbSNP, Human Build 132, http://www.ncbi.nlm.nih.gov/projects/SNP/ GeneNote, http://bioinfo2.weizmann.ac.il/cgi-bin/genenote/ home_page.pl and 17 GRCh37 patch 2, obtained from ftp://ftp.ensembl.org/pub/release-61/ NHLBI Exome Sequencing Project, http://evs.gs.washington. edu/EVS/ Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/ Panther, http://www.pantherdb.org/tools/csnpScoreForm.jsp/ PMut, http://mmb.pcb.ub.es/PMut/ SNAP, https://www.rostlab.org/services/snap/ SNPs&GO, http://snps-and-go.biocomp.unibo.it/snps-and-go/ UniGene, http://www.ncbi.nlm. nih.gov/unigene/ 18 References 1. Murphy, S.M., Laura, M., Fawcett, K., Pandraud, A., Liu, Y.T., Davidson, G.L., Rossor, A.M., Polke, J.M., Castleman, V., Manji, H., et al. (2012). Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. Journal of neurology, neurosurgery, and psychiatry 83, 706-710. 2. Saporta, A.S., Sottile, S.L., Miller, L.J., Feely, S.M., Siskind, C.E., and Shy, M.E. (2011). Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol 69, 22-33. 3. Rossor, A.M., Polke, J.M., Houlden, H., and Reilly, M.M. (2013). Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nature reviews Neurology 9, 562-571. 4. Harding, A.E., and Thomas, P.K. (1980). The clinical features of hereditary motor and sensory neuropathy types I and II. Brain : a journal of neurology 103, 259-280. 5. Lin, K.P., Soong, B.W., Yang, C.C., Huang, L.W., Chang, M.H., Lee, I.H., Antonellis, A., and Lee, Y.C. (2011). The mutational spectrum in a cohort of Charcot-Marie-Tooth disease type 2 among the Han Chinese in Taiwan. PloS one 6, e29393. 6. Soong, B.W., Huang, Y.H., Tsai, P.C., Huang, C.C., Pan, H.C., Lu, Y.C., Chien, H.J., Liu, T.T., Chang, M.H., Lin, K.P., et al. (2013). Exome sequencing identifies GNB4 mutations as a cause of dominant intermediate Charcot-Marie-Tooth disease. American journal of human genetics 92, 422-430. 7. Genomes Project, C., Abecasis, G.R., Altshuler, D., Auton, A., Brooks, L.D., Durbin, R.M., Gibbs, R.A., Hurles, M.E., and McVean, G.A. (2010). A map of human genome variation from population-scale sequencing. Nature 467, 1061-1073. 8. Calabrese, R., Capriotti, E., Fariselli, P., Martelli, P.L., and Casadio, R. (2009). Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum Mutat 30, 1237-1244. 9. Thomas, P.D., Campbell, M.J., Kejariwal, A., Mi, H., Karlak, B., Daverman, R., Diemer, K., Muruganujan, A., and Narechania, A. (2003). PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13, 2129-2141. 10. Ferrer-Costa, C., Gelpi, J.L., Zamakola, L., Parraga, I., de la Cruz, X., and Orozco, M. (2005). PMUT: a web-based tool for the annotation of pathological 19 mutations on proteins. Bioinformatics 21, 3176-3178. 11. Bromberg, Y., and Rost, B. (2007). SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic acids research 35, 3823-3835. 12. Greco, A., Mariani, C., Miranda, C., Lupas, A., Pagliardini, S., Pomati, M., and Pierotti, M.A. (1995). The DNA rearrangement that generates the TRK-T3 oncogene involves a novel gene on chromosome 3 whose product has a potential coiled-coil domain. Molecular and cellular biology 15, 6118-6127. 13. Hernandez, L., Pinyol, M., Hernandez, S., Bea, S., Pulford, K., Rosenwald, A., Lamant, L., Falini, B., Ott, G., Mason, D.Y., et al. (1999). TRK-fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations. Blood 94, 3265-3268. 14. Hisaoka, M., Ishida, T., Imamura, T., and Hashimoto, H. (2004). TFG is a novel fusion partner of NOR1 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer 40, 325-328. 15. Ishiura, H., Sako, W., Yoshida, M., Kawarai, T., Tanabe, O., Goto, J., Takahashi, Y., Date, H., Mitsui, J., Ahsan, B., et al. (2012). The TRK-fused gene is mutated in hereditary motor and sensory neuropathy with proximal dominant involvement. American journal of human genetics 91, 320-329. 16. Witte, K., Schuh, A.L., Hegermann, J., Sarkeshik, A., Mayers, J.R., Schwarze, K., Yates, J.R., 3rd, Eimer, S., and Audhya, A. (2011). TFG-1 function in protein secretion and oncogenesis. Nat Cell Biol 13, 550-558. 17. Beetz, C., Johnson, A., Schuh, A.L., Thakur, S., Varga, R.E., Fothergill, T., Hertel, N., Bomba-Warczak, E., Thiele, H., Nurnberg, G., et al. (2013). Inhibition of TFG function causes hereditary axon degeneration by impairing endoplasmic reticulum structure. Proc Natl Acad Sci U S A 110, 5091-5096. 18. Lee, S.S., Lee, H.J., Park, J.M., Hong, Y.B., Park, K.D., Yoo, J.H., Koo, H., Jung, S.C., Park, H.S., Lee, J.H., et al. (2013). Proximal dominant hereditary motor and sensory neuropathy with proximal dominance association with mutation in the TRK-fused gene. JAMA Neurol 70, 607-615. 19. Wang, Y., Bruce, A.T., Tu, C., Ma, K., Zeng, L., Zheng, P., Liu, Y., and Liu, Y. (2011). Protein aggregation of SERCA2 mutants associated with Darier disease elicits ER stress and apoptosis in keratinocytes. Journal of cell science 124, 3568-3580. 20. Conchillo-Sole, O., de Groot, N.S., Aviles, F.X., Vendrell, J., Daura, X., and Ventura, S. (2007). AGGRESCAN: a server for the prediction and evaluation of "hot spots" of aggregation in polypeptides. BMC bioinformatics 8, 65. 21. Zhang, K., and Kaufman, R.J. (2004). Signaling the unfolded protein response from the endoplasmic reticulum. The Journal of biological chemistry 279, 20 25935-25938. 22. Li, F., Hayashi, T., Jin, G., Deguchi, K., Nagotani, S., Nagano, I., Shoji, M., Chan, P.H., and Abe, K. (2005). The protective effect of dantrolene on ischemic neuronal cell death is associated with reduced expression of endoplasmic reticulum stress markers. Brain research 1048, 59-68. 23. Hirota, M., Kitagaki, M., Itagaki, H., and Aiba, S. (2006). Quantitative measurement of spliced XBP1 mRNA as an indicator of endoplasmic reticulum stress. The Journal of toxicological sciences 31, 149-156. 24. Saporta, M.A., Shy, B.R., Patzko, A., Bai, Y., Pennuto, M., Ferri, C., Tinelli, E., Saveri, P., Kirschner, D., Crowther, M., et al. (2012). MpzR98C arrests Schwann cell development in a mouse model of early-onset Charcot-Marie-Tooth disease type 1B. Brain : a journal of neurology 135, 2032-2047. 25. Badr, C.E., Hewett, J.W., Breakefield, X.O., and Tannous, B.A. (2007). A highly sensitive assay for monitoring the secretory pathway and ER stress. PloS one 2, e571. 26. Valenzuela, J.I., Jaureguiberry-Bravo, M., and Couve, A. (2011). Neuronal protein trafficking: emerging consequences of endoplasmic reticulum dynamics. Molecular and cellular neurosciences 48, 269-277. 27. Aridor, M., and Fish, K.N. (2009). Selective targeting of ER exit sites supports axon development. Traffic 10, 1669-1684. 28. Millecamps, S., and Julien, J.P. (2013). Axonal transport deficits and neurodegenerative diseases. Nature reviews Neuroscience 14, 161-176.