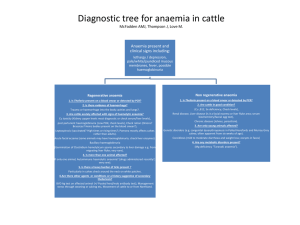
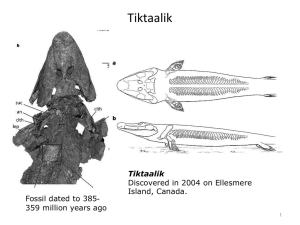
Haem Module 3
advertisement

3.1 – GLOBIN GENES: STRUCTURE AND FUNCTION 1. Importance of globin genes encode for proteins which make up haemoglobin – essential physiologically prevalent monogenic disorders commonly affect globin genes their evolution are paradigms for other vertebrate genes 2. Structure of Haemoglobin & types of globin chains Haemoglobin 65.4kDa protein that constitutes 90% of RBC protein o CA is next biggest protein constituent made of 2a-like and 2b-like globin chains, which are roughly 50% similar each globin chain is associated with a porphyrin ring, with a haem group and Fe atom in its centre o The porphyrin ring binds to histidine residues on the E7 and F8 domains of globin o oxygen lies between the E7 histidine and iron atom oxygen binding subtly changes the conformation of haemoglobin from a tight protein (T) to a relaxed one (R) o binding one O2 makes binding another easier. the same for losing one O2 Types of Globin & types of Haemoglobin A-like alpha2 alpha2 alpha2 zeta2 zeta2 alpha2 b-like beta2 delta2 gamma2 gamm2 epsilon2 epsilon2 Hb name HbA HbA2 HbF Hb Portland Hb Gower1 HbGower2 description Adult; 97% Adult; 1-3% Fetal; also <1% of total adult Hb Embryonic 3. Globin gene & temporal relationship to haemoglobin subtypes The alpha-like cluster lies on chromosome 16 o there are two alpha genes downstream of the embryonic zeta gene The beta-like cluster lies on chromosome 11 o there are 2 gamma genes and one each of epsilon, delta and beta transcription of the genes changes temporally in relation to their distance from the 5’ end o except for delta, which lies 5’ to beta but is probably transcribed later in life embryonic transcription occurs in the yolk sac fetal transcription occurs in the liver and spleen; adult transcription occurs in the BM all genes are highly conserved – 3 exons and 2 introns 4. brief evolution of globin genes universal monogenic globin gene underwent duplication and mutation both alpha and beta on the same chromosome o this occurs in meiosis due to unequal recominbation transposition led to separation of the genes alpha and beta then gave rise to other variants and pseudogenes (relics) 5. molecular pathology in thalassaemias beta-thalassaemia nonsense mutations o this is where a coding AA is replaced by a stop codon following mutation frameshift mutations o this is where 2 adjacent bases are deleted = shift in reading frame defective mRNA splicing o splice junction mutations = failure of splicing = translation of introns (particularly problematic if introns contain stop codons; subsequent exons will not be transcribed) o cryptic mutations = mutation within an intron that causes splicing machinery to use a different splice-acceptor-site eg GA at 110 on intron 1; mediterrarean variant associated with severe disease promoter region/ polyA mutations o promoter mutations associated with decreased mRNA production; mild disease deletions alpha-thalassaemia deletions are a more prominent feature because one defective allele following point mutation can usually be compensated for by the other 3 (see alpha thal lecture) 6. regulation of the beta globin locus Challenges to regulatory mechanisms the gene must be tissue (erythroid) specific and therefore so must regulatory mechanisms you need to make the right absolute and relative (to alpha) amounts of beta globin you need to make sure to can regulate all types of globin and accommodate for switching Regulation Locus control region; has 7 hypersensitive sites to DNAases and influences all b-like genes o confers position and copy number dependence, as well as erythroid specificity because they have binding sites for erythroid specific TFs (see below) individual genes have their own promoter/enhancer regions o promoters work in cis and are always 5’ to the one gene they control o enhancers work at a distance and can be 5’ or 3’ to the gene they control transcription factors o erythroid TFs include GATA1, EKLF, NFE2, SSP o ubiquitous TFs (in all cells) include YY1, SP1, AP1, USF o TF binding sites (LCR, promoter/enhancer regions) are highly conserved. example sequences are CACCC, CCAAT, TATA. chromatin structure & histone modifications o (open) euchromatin is transcribed more readily. methylation inhibits transcription o histone acetylation promotes transcription 7. methods to study globin genes & globin gene regulation study the effects of prevalent mutations of DNA amongst disease sufferers analyse natural mutations in humans o eg the gamma promoter region was found to be mutated in hereditary persistence of HbF disease transgenic technology applied to mice o standard transgenics putting segments of human DNA into mice and seeing is they make the human protein o knockout studies GATA1 deficient mice demonstrate erythroid maturation block at the proerythroblast stage EKLF deficient mice die in utero and demonstrate globin chain imabalance o Highly conserved regions MUST be important for gene regulation; some key regulatory regions for alpha genes are highly conserved regions within introns of other genes The thinking is that conserved regions are the binding sites for TFs Modelling o competitive model of gene switching fetal and adult globin genes compete for the interaction of the LCR. The transcriptional environment favors particular interactions and obviously the environment changes following birth. FKLF and SSP important for HbF (delta); EKLF for HbA (beta) o autonomous silencing model opposite of competition; suggests that genes get progressively silenced Sequences necessary for silencing of e and g genes have been mapped to their 5’ proximal regions (eg CACCC box in g gene) 3.2 – HAEMOGLOBIN STRUCTURE AND FUNCTION 1. synthesis of Haem 1st stage happens in mitochondria, second in cytosol and third in mitochondria again. iron needed for first step; stimulates ALA synthase haem takes part in auto-negative feedback by inhibiting its own synthesis (ferrochelatase inhibition) and iron uptake Ala placed in urine cup produces heme 2. Functions of Haemoglobin O2 Transport o 2,3DPG favours O2 release (and is often upregulated in anaemia to do this) o the O2 disso curve is due to cooperativity o 2,3DPG, temperature, acidity and HbS all shift the curve right CO2 Transport o although most CO2 is transported dissolved in the plasma, about 15% binds to the nterminus of a-globin in deoxyhaemoglobin; forming a carbamate ion COO Buffering Nitric oxide scavenging/ transport (3 functions) o a) converts NO to nitrate inside RBCs; free Hb in the plasma also does this oxyHb + NO + O2 + 2H+ metHb + nitrate + H2O hence excess IV haemolysis = free Hb = less NO = vasoconstriction o b)converts nitrite to nitrate oxyHb + nitrite +O2+2H+ metHb + Nitrate + H2O o c) when there is a lack of oxygen (in tissues), deoxyHb regenerates NO from nitrite 3. mutations in globin genes May not have any effect (synonymous/same-sense mutation) may encode a different AA with no functional abnormality may encode a different AA with a resulting functional abnormality (see below) change a coding sequence to a stop codon and give a shorter transcript change a stop codon to a coding one and give a larger transcript o (either change in size can affect stability) 4. functional abnormalities resulting from globin gene mutation Polymerisation of Hb at low O2 pressure and decreased O2 affinity (HbS) mutations affect ab2 bonds or favour formation of deoxyHb Reduced Hb solubility (HbC) HbC heterozygosity is prevalent in western Africa (malaria protection) heterozygosity is harmless; blood film often shows boat, target cells and IRCs homozygosity leads to crystillation of HbC, cellular dehydration and rigidity and intravascular haemolysis o = anaemia, hyperbilirubinaemia, gallstones increased O2 affinity (Hb Luton, Hb Kempsey) a1b2 bond mutations = defective cooperativity (hindered movement/rotation) curve shifts left = reduced O2 delivery to tissues = EPO response & polycythaemia when HCT exceed 0.5, increased blood viscosity is another problem carboxyHb (high in smokers) also interrupts cooperativity and shifts curve left. Hb instability (Hb Koln) mutations in histidine or a1b1 bonds leads to haem falling out of porphyrin in favour of water. if damage is extensive enough, the tetramer may dissociate the precipitation/ loss of haem is stimulated by infection/ oxidant drugs haem-less Hb precipitates as Heinz bodies and increases cell rigidity = haemolysis Hb Koln may demonstrate blister cells, where a portion of the cell appears empty heat instability test may diagnose other mutations generally affect crucial sites eg a1b2 interface, c terminal of beta chain, mutations in 2-3dpg binding site Tendency for Hb to oxidise (Hb M; Hyde Park) In HbM - mutations replace histidine with tyrosine so that iron is stable as Fe3+ = inability to transport O2 = anaemia absorption spectra are similar to that of methaemoglobin production of a Hb that interacts adversely with HbS C, Oarab, Dpunjub HbCHbS is not as severe as SCA because HbC has a normal O2 affinity so blood Hb is higher/normal o SC poikilocytes are curved cells (S polymerisation) with rectiangular protrusions (C crystals) Production of a (thalassaemic) Hb at a reduced rate which can thus interact with thalassaemias deletion of a LCR gene deletion (delta.beta thalassaemia) gene fusion (Lepore) promoter sequence/ initiation codon mutation change in gene length RNA processing (splicing/ capping/ tailing) mutations - HbE 5. Examples of variant haemoblogins with more than one abnormality Polyumerisation and low O2 affinity (HbS) unstable with tendency to oxidise/ change in affinity methaemoglobinaemia with change in affinity 3.3 – SICKLE CELL DISEASE 1. Genetic basis of SCD codon 6 = change from GAG to GTG = swapping glutamic acid for valine (uncharged) 2. Mechanisms in reversible sickling Role of valine fits into a hydrophobic pocket of neighbouring deoxyHbS triggers polymerisation of deoxyHbS and formation of 7 twisted double fibres of HbS o HbA and Heterotetramers (alpha2, beta, betaS) have a reduced ability to form polymers because normal beta chains can’t engage with the hydrophobic pocket Polymerisation deform the cell into a sickle shape. Polymerisation is triggered by o hypoxia o infection (low pH, increased temperature, increased OSM) Sickling is initially reversible (‘boat cell’), but ultimately becomes irreversible o we know it is a reversible process because veins have more sickled cells than arteries o sickling manifests in organs where there is a long time for cells to travel from the arterial bed to the venous bed. this allows time for sickling. the spleen is an example o secondary events (see below) makes cells irreversibly sickled 3. Mechanisms in irreversible sickling Main mechanisms – increased adhesion, increased rigidity and change in cell shape Further roles of HbS metHbS is formed via oxidation. it breaks down into hemichromes and releases haem and Fe3+ Haem and Fe3+ oxidise membrane lipids, cytoskeleton and other HbS molecules oxidised HbS precipitates as Heinz bodies which bind to ankyrin band 3 on inner CSM o this is seen and removed by the spleen; extravascular haemolysis Cytoskeletal damage Oxidation = loss of ability to tether to CSM = loss of CSM vesicles decreased CSM:cell content ratio means that cell becomes more rigid CSM damage/loss Polymers of HbS can pierce the membrane/ produce a spicule which is then lost oxidation of lipids = rearrangement of inner and outer CSM lipids and loss of lipids o relocation of PPDserine to outer CSM = increased adhesion to endothelium & macrophages (this happens before changes in cell shape and rigidity) phagocytosis by macrophages complement activation = intravascular haemolysis prothrombotic effect increased IgG binding = intravascular haemolysis Cellular dehydration (high MCHC) Oxidative damage to CSM = activation of ion transport channels = loss of K + Cl = loss of water Mg usually inhibits KCl cotransport; SCD patients usually have low Mg Polymerisation increases CSM permeability = Ca influx & activation of Gardos channels Roles of other cells Endothelial dysfunction = prothrombotic, increased adhesion, and production of pro inflammatory cytokines o pro inflammatory cytokines also activate Gardos channels Macrophages bind to Ig and complement on RBCs, particularly those with altered CSMs, and phagocytose them Neutrophils are activated, particularly during infection, and this increases their adhesive properties o adhesion of neutrophils to endothelium helps to trap RBCs in venules = increased transit time = promoted sickling Role of NO IV haemolysis = free Hb in circulation = breakdown of NO = vasoconstriction and more platelet activation o haptoblobin and haemopexin, two mechanisms by which Free Hb is removed from the blood, are both reduced in SCD o IV haemolysis = release of arginase = breakdown of arginine = less NO synthesis activated neutrophils (infection) also inhibit eNOS = decreased synthesis Low NO correlates with high LDH (infarcts) 4. Pathogenesis of Vaso-occlusion in SCD VCAM1 is upregulated by the dysfunctional endothelium. it binds to reticulocytes via VLA4 reversibly sickled cells also bind to dysfunctional endothelium and exposed ECM o young sickle cells bind to integrins via LW antigen. LW antigen is upregulated by adrenaline so stress/adrenaline = adhesion and may lead to sickling The majority of adhesion is believed to occur in post-capillary venules o RBCs elongate in capillary bed (surface area) and there is slow blood flow in the venule o adhesion leads to retrograde capillary obstruction = ischaemia/infarction Vaso-occlusive disease starts after 6 months of life – HbF protects upto this point o Painful crises (limbs/chest/abdomen) occur in 40% of patients? o hand foot syndrome (dactylitis) in the first two years of life leads to growth delay o adolescent sequelae include delayed puberty and priapism 5. Pathogenesis of stroke in SCD Years 1-10 ischaemic stroke due to microinfarcts (17% are not apparent clinically) haemorrhagic stroke o endothelial dysfunction = intimal (fibroblasts/SMC) hyperplasia = narrowed carotids etc = aneurysms = haemorrhage o narrowed carotids give turbulent blood flow; Doppler monitoring and transfusions can help prevent stroke (adams 1998) 6. Pathogenesis of ACS in SCD Years 3 microthrombi in pulmonary vessels = lung infarcts vasoconstriction due to less NO due to free Hb is another important mechanism rib infarcts = less deep breathing (pain) = hypoxia = sickling importantly, ACS hypoxia sickling ACS o other things that make ACS worse include infection and BM emboli clinical features include fever, CP, cough, SOB, wheeze, cyanosis and LOC due to hypoxia 7. Splenic dysfunction in SCD Haemolysis Splenomegaly recurrent sequestration crises due to infarcts atrophy and fibrosis of spleen hyposplenism (Howel Jolly bodies) recurrent infections occur due to immune dysfunction, particularly malaria, pneumococcus and meningococcus splenomegaly usually disappears after 5 years. persistent splenomegaly may indicate thalassaemia or higher HbF 8. Pathogenesis of Pulmonary HTN in SCD recurrent Episodes of ACS can lead to this because it causes thrombosis, vasoconstriction and thickening of vascular walls/fibrosis o Low NO (free Hb) also contributes to thrombosis and vasoconstriction can lead to right heart failure and death 9. pathogenesis of priapism in SCD priapism is an unwanted, painful, persistent erection (acute emergency) Poor venous drainage from corpus spongiosum is the cause o decreased NO synthesis (arginase release from RBCs) = vasoconstriction o free Hb = NO scavenging o Increased reticulocyte adhesion = slow flow = sickling 10. pathogenesis of bone disease in SCD Bone infarcts lead to a spectrum of disease o osteomyeleitis (particularly staphylococcus and salmonella) o infarct = osteonecrosis chronic osteoarthritis o infarct = BM emboli, particularly to lungs 11. renal dysfunction in SCD Increased OSM + hypoxia infarcts haematuria, papillary necrosis, loss of concentrating ability o hence pay particular attention for dehydration in SCD sickle glomerulopathy CRF & Nephrotic syndrome CRF recurrent infections 12. retinal dysfunction in SCD vascular obstruction vitreous haemorrhage. HbSHbC is particularly associated because HbC has a normal O2 affinity so you have higher Hb = viscosity 13. Why does SCD cause anaemia? red cell life is reduced 10 fold due to intra- and extravascular haemolysis EPO response means that erythropoiesis is increases 3-6fold = increase in plasma volume o expansion of BM increases need for folic acid. folic acid deficiency can contribute to anaemia in Africa HbS has a lower affinity for O2 in the first place – this impairs the EPO response parvovirus B19 infection arrests haematopoiesis and can lead to symptomatic aplastic crises 14. why does SCD cause thrombosis? endothelial activation, adhesion and increased TF activity Platelet activation due to less NO CSM changes (externalisation of PPDserine) is prothrombotic reduction of Protein C and protein S increased prevalence of APS antibodies 15. Why does SCD cause infection? Hyposplenism, osteomyelities, cholecystitis, leg ulcers and renal infarcts 16. interactions of HbS with other variant haemoglobins alpha-thalassaemia trait Higher HbA2 percentage = lower HbS concentration = less sickling and haemolysis, O2 affinity increased, higher Hb o less sickling may preserve flow small vessels; stroke is less frequent and splenic function is better preserved higher Hb = increased viscosity so vaso-occlusive sequelae in large vessels are more frequent than in SCD o particularly painful crises, avascular necrosis and osteonecrosis HbC very similar genetic mutation to HbS; mutation from GAG AAG in 6th codon of beta globin swap from glutamic acid to lysine HbC binds to K+Cl- cotransporter and aggravates cellular dehydration = ICCs o contracted cell = increased HbS conc = sickling HbC has greater O2 affinity = higher Hb conc = small vessel thrombotic risk (retinopathy increases 10x) less sickling = delay in splenic atrophy/fibrosis = more common splenic crises HbOarab also does this and may also activate Gardos channels More severe than SCD Similar to SCD Milder than SCD Considerably milder than SCD HbSHbSantilles HbS/B0thal, S/C.harlem, S/Oarab S/C, S/Dpunjab, S/B+thal, S/lepore S/B++thal, S/DBthal, S/E, S/persistence of HbF 3.4 – VARIANT HAEMOGLOBINS (remember to look at HPLC stuff from intro) 1. introduction to variant haemoglobins most mutations that lead to formation of variant Hb are single AA replacements o others include fusion Hbs, deletions, changes in size mutations may affect one/more of structure, function, production rate and stability only a few variants are clinically significant and lead to: o sickling – AS/SS/SC/SDpunjab/SOarab/SBthal o thalassaemias – beta – Lepore (fusion), HbE (mRNA processing mutations) alpha – Constant spring (short chain), quong Sze (unstable) o unstable haemoglobins o altered O2 affinity o M haemoglobins 2. detection of variant haemoglobins Clinical presentation o anaemia, erythrocytosis, cyanosis, haemolyisis Pre-op/antenatal/neonatal haemoglobinopathy screening o FBC indices – Hb, RBC, MCV. MCH, MCHC, RDW morphology - microcytic anaemia, hypochromia, anisocytosis target/irregularly contracted cells in HbC basophilic stippling may point out unstable Hbs Heinz bodies/ pappenheimer bodies o sickle solubility positive result when HbS>20% o Hb separation studies (electrophoresis, HPLC) HPLC now the gold standard in countries that can afford it o antibody detection for haemolytic anaemias 3. beta globin variants apart from HbS HbE o change from 26th AA from glutamic acid lysine; leads to reduced mRNA production o The mutation is located near to the junction between exon1 and intron1 and it unmasks a cryptic splice site, which is not normally used. this site competes with the normal splice site. Some mRNAs are correctly processed however, a large amount of mRNA is wrongly processed and reaches an early stop codon. o trait is prevalent in N. India and SE asia o homozygosity and carrier status have a mild microcytic anaemia o need to rule out compound heterozygosity with B0thal – which resembles b.thal o travels with C on alkaline electrophoresis and A on acid electrophoresis o lies in the A2 band on HPLC; suspect if A2 rises to 25-30% HbC o change from 6th AA from glutamic acid lysine o trait is prevalent in west Africa o heterozygotes are largely asymptomatic; homozygotes have a mild haemolytic (hyperchromic, high MCHC) anaemia, splenomegaly; look for target cells and ICCs HbD o change from 121st AA from glutamic acid glutamine o carrier status common in Punjab/ los angeles o target cells abundant HbO o change from 121st AA from glutamic acid to lysine o prevalent in north Africa and middle east (arab) o compound heterozygosity with HbS is similar to SCA Hb Lepore o fusion of delta(1-87) and beta (116-146) following erroneous recombination in meiosis o particularly prevalent in middle and eastern Europe o homozygosity is similar to B-thal major HbH o loss of three alpha genes leads to 4 beta chains composing the Hb tetramer o can be asymptomatic or can result in severe anaemia dependent on transfusions o teardrop cells in blood film may be suggestive; look for precipitating granules after new methylene blue staining as well o fast moving on electrophoresis o low HbA2 HbKoln o change from 98th AA from valine methionine makes the molecule unstable o instability = reduced life span = haemolytic anaemia appearing from 6months onwards as beta globin synthesis starts o Film = Heinz bodies, hypochromic RBCs on film, methaemoglobinaemia o indices = mild anaemia and reticulocytosis out of proportion with that anaemia o falls in HbA window on HPLC 4. Other globin variants Delta globin variants Variant HbA2 (change from 16th AA from glycine arginine) and Hb Lepore (see above gamma globin variants HbBarts o loss of 4 alpha genes means where 4 gamma globins make up the Hb tetramer o fast moving on electrophoresis o can result in hydrops fetalis without transfusions In utero HbPoole o change from 130th AA trom trp glycine makes the molecule unstable o presents with haemolytic anaemia in childhood that diasappears as gamm globin synthesis ceases by end of first year of life alpha chain variants o HbG change from 68th AA from asparagine lysine most commonly found in black Americans (Philadelphia) split HbA2 reading on HPLC High Performance Liquid chromatography Percentage of Hb in sample is demonstrated on a graph based on their retention time Furthest left is Injection artifact (and bilirubin if it is a haemolytic anaemia sample) then, in order, is HbF, A, A2 & S o each Hb will have, in order, glycosylated (or acetylated for HbF) and post-translation modified product peaks prior to it; these peaks always exist and may be hidden in others 3.5 – BETA THALASSAEMIAS 1. introduction to thalassaemia thalassaemia = reduced rate of synthesis of one more globin chains, and thus haemoglobin. usually produces a microcytic anaemia any globin gene can be affected, and more than one can be affected at the same time alpha and beta thalassaemia are the clinically important thalassaemias alpha globin is a component of most haemoglobins except some embryonic forms, so alpha thalassaemia, unlike beta, is symptomatic from fetal life onwards (beta from 1 year onwards) in beta thalassaemia: o HbA is not formed/ formed at a reduced rate (no/less beta globin) o HbA2 rises upto 10% as compensation and HbF may also do this (neither have b globin) o High HbA2 is useful in the diagnosis of beta-thal no diagnostic use in neonates; delta globin synthesis starts at 30weeks 2. genetic syndromes arising beta thalassaemia Reduced rate of synthesis = B+ o a very mildly reduced rate = B++ absent synthesis = B0 BB B+B B0B B+B+ B0B0 B+B0 Normal heterozygosity/ trait homozygosity these may not be true homozygotes because each allele may still have a different mutation – compound hetero compound heterozygoisty 3. Mutations in beta thalassaemia MAINLY POINT MUTATIONS affecting: o promoter region o mRNA processing: splice site (+cryptic splice mutations), CAP/PolyA site o size of transcript (coding AA for stop codon and vice versa); can lead to instability Deletions in entire gene or LCR region o mutations that result in a variant thalassaemic Hb: o eg HbE or HbLepore (delta-beta fusion), epsilon-gamma-delta-beta Hb; (similar to b-thal trait but difficult to diagnose because delta mutation means that A2 will not be raised) 4. clinical syndromes arising in beta thalassaemia Beta thalassaemia major – transfusion is essential o usually associated with homozygous/compound heterozygous genotypes beta thalassaemia intermedia – heterogenous presentation where survival is possible without transfusion o extramedullary haemopoiesis, leg ulcers, gallstones, hypercoagulability and pulm HTN can all be present. o Bad anaemia may stimulate Fe absorption = iron overload o heterogenous basis of geneotype Homozygosity but protection from a major: mutated genes were both mutated mildly coexisting alpha thal normalizes the a:b ratio coinheritance of hereditary persistence of HbF Heterozygosity but worsening of a minor the mutated allele is severely mutated (dominant mutation) coexisting alpha chain excess worsens the ratio beta thalassaemia minor – asymptomatic o usually associated with heterozygous/trait status o heterozygosity = unbalanced chain synthesis = increased a:b ratio o diagnosis is based on indices, film, increased HbA2 in the absence of symptoms Indices = almost normal Hb, low MCV, low MCH, high RBC (EPO response) Film = hypochromia, microcytes, some pikilocytes o symptoms may arise in infection/pregnancy- but the main concern with these patients is giving birth to a beta-thal major child (25%); screening 5. clinical features of beta thalassaemia major Major pathology is severe imbalance in globin chains o reduced beta chain directly leads to anaemia because of decreased Hb synthesis o increased alpha chains precipitate = damage to erythroid precursors = ineffective EPO response = marrow hyperplasia o damage to eryhtroid precursors contributes to haemolysis = anaemia o marrow/ eryhtroid hyperplasia = bone deformities eg osteoporosis ‘hair on end’ appearance of skull on XR Since BM cannot effectively respond to anaemia, extramedullary erythropoiesis is triggered o splenomegaly pooling of RBCs hypersplenism o hepatomegaly general appearance = abdominal enlargement, respiratory distress (ribs drawn in), wasted limbs, thick skull/jaw Diagnosis/Investigation Indices - microcytic anaemia Film o pappenheimer bodies = iron granules in RBCs o a-chain precipitates in RBCs o some nucleated red cells as evidence of erythroid hyperplasia o erythroblasts may appear devoid of Hb +/- vacuoles, lobulated nuclei, basophilic stippling HPLC – absence of HbA, high HBA2 (and HbF sometimes) Management Conservative – PREVENTING THE MAJOR CHILD BEING BORN o ante- & prenatal sreening, with the option of termination o model 1980 lancet said abortions are now down from 70% to 30% as a result of couples just not taking the risk Medical - principle of treatment is monthly transfusion from infancy the major risk is iron overload – particularly affectin heart, liver and endocrine glands o Cardiac Fe overload accounts for 75% of beta-thal deaths in Sardinia (galanello 2003) o iron chelation therapy (SC infusion 5-6days/week) with desferrioxamine helps o compliance (social problem) determines survival (thalassaemia international federation) hydroxycarbamide helps to stimulate HbF synthesis application of treatment is poor in N.Africa, middle east and SE asia o transfusions are a sparse resource o screening/ chelating treatment is expensive and it is prevalent in poor countries o cultural problems – muslims are less compliant? those who don’t accept abortion. those who will have the child anyway (poor people, workers on a farm etc) 3.6 – INTERACTIONS OF BETA THALASSAEMIA 1. Compound heterozygous states that are clinically significant o When the other allele codes for a globin which is dangerous in high concs o S, C, unstable alleles o this is because you end up being totally unable to produce HbA o when the other allele codes for a variant ‘thalassaemic’ haemoglobin o E, Lepore o When two different carriers reproduce, there is a 25% chance each of the offspring being normal, either heterozygote or a compound heterozygote 2. Compund heterozygooisty with HbS o is a type of SCD o but much rarer than SCA and HbSHbC o HbS/Hbthal+ is milder than HbS/Hb0 because you make more HbA; disrupts polymer formation o both forms are milder anaemias than SCA because of higher levels of HbA2 and HbF Increased HbA2 = increased viscosity and less sickling osteonecrosis increased by 30% compared to SCD 3. Compund heterozygooisty with HbC o Not a clinical problem o blood film might show features of thalassaemia alongside HbC crystals and target cells 4. Compund heterozygooisty with HbE o HbE only contributes 25% to the compound instead of 50%, hence this usually presents with thalassaemic properties (microcytic anaemia and thalassaemic indices) o often comparable to a thalassaemia minor or intermedia, but severity is not easily predictable o since HbE is a thalassaemic Hb the net result is: o very low globin chain and hence Hb synthesis = anaemia o high alpha:beta ratio = precipitates = ineffective erythropoiesis o compound heterozygopoisty accounts for a lot of beta-thalassaemia in SE asia 3.7 - EPIDEMIOLOGY OF RED BLOOD CELL DISORDERS/ MALARIA 1. To understand the hypothesis of gene selection in terms of survival advantage as applied to malaria Immigrant populations in the US have a high frequency of haemoglobinopathies and thalassaemias. Reasons for this could be: o Spontaneous mutation in this group, giving rise to the disease syndromes o The disease syndromes conferred a survival advantage (malaria) Haldane 1949 observed that The corpuscules of anaemic heterozygotes (for thalassaemia) are smaller than normal, and more resistant to hypotonic solutions; possibly conferring resistance to malaria Impact of malaria: o Incidence 350-500million; 41% of world population lives in endemic areas o Mortality upto 25-30%; 1.5mill deaths/year mostly in children Red cell disorders that are protective against malaria include: o Blood group antigen - Duffy/ Glycophorin variants o Red cell membrane - Ellyptocytosis/ ovalocytosis o Hb disorders - Haemoglobinopathies/ variants/ Thalassaemias o Enzyme disorders - G-6-PD deficiency Since the traits that are protective are so heterogeneous, there are probably multiple mechanisms in protection, and polymorphisms may even be evolving The protective effects have been demonstrated by o Monozygotic twin studies o Family studies o Observing ethnic differences of disease prevalence within the same environment Eg sub-sharan Africa (HbS) have resistance to plasmodium vivax Eg Nepalese people with a-thal have lower malaria rates than those without Eg Fulani tribe west Africa (protective antibodies) have low malaria rates o Genetic approach using case control/ epidemiological studies 2. To understand how malaria is likely to have altered the prevalence of red blood cell disorders HbS HbS prevalent in Africa and parts of south eastern Europe Sickling disorders have high childhood mortality (infection), reduced life expectancy on average and increased hospital demands/drain on resources SC trait affects about 29% of Nigerians by age 5, there is almost no SCA at this age o SCA is usually lethal in childhood. ; it has higher rates of parasitaemia than HbA o Normal HbA has higher frequency and density of parasitaemia that SC trait SC trait gives 92% reduction in relative risk of severe malaria in one case control study o 10 fold increased protection against severe malaria but not mild malaria o Protection increases with age suggesting enhanced immunity In vitro studies suggest SC trait and SCA both give: o Impaired parasite growth Sickling, dehydration and haemolysis of parasitised cells Polymerised HbS either poor substrate/ damages parasite function by ‘impaling’ o o Slow growth rate in low oxygen tension Increased removal by Reticuloendothelial cells Sickling gives enhanced immune recognition Impaired rosetting (blood group O also does this) HbC HbC prevalent in west Africa Carriers and homozygotes are both protected. Possible mechanisms are: o Reduced cytoadherence o Abnormal expression of surface protein PfEMP1 o Clustering of band 3 HbE HbE prevalent in N.India and SE.Asia Mechanisms of protection may include resistance to parasite invasion and enhanced clearance Alpha-thalassaemia 80% of the tharu Nepalese have alpha-thal and have lower malaria rates o Alpha-thal distribution amongst these people altitude (malaria) and latitude dependant o Genetic linkage analyses showed that this is not due to founder effect o Early exposure to vivax may boost immunity to falciparum leading to reduced morbidity o May also be protective against other infections The mechanism of protection is unknown but may include: o Reduced parasite growth o Increased antibody binding o Enhanced splenic clearance Duffy antigen (Fy)variants thought to be receptor for P. vivax; most black Africans lack duffy antigen Blacks from West Africa as well as US rarely have P.vivax malaria o Resistance to P. knowlsei has been deomstrated in vitro It is thought that other CSM variants (glycophorin) only partially reduce parasitic invasion 3. To understand the basic mechanisms of how red cell and other genetic polymorphisms can protect against the severe effects of malaria A balanced polymorphism is deleterious in the homozygous state but protective in the heterozygous state o i.e over time (and the time period required depends on the degree on protection), the compensation of the heterozygous state for the deleterious effect that would otherwise be conferred by the homozygous state, becomes fixed/universal Some non-RBC polymorphisms that maybe interact with malaria: o HLA - B53 and DRB*1302 = lower risk o TNF secretion promotion - polymorphism of promoter 308 = higher risk o ICAM1, iNOS promoter and Haptoglobin polymoprhisms also implicated Impact of genetic red cell disorders: o becoming a global problem with major implications on healthcare resources o Treatment is usually expensive and not available in poor countries o There is need for increased data o We need to target resources, education, pre natal diagnosis 3.8 - ALPHA THALASSAEMIA 1. Introduction to alpha thalassaemia Alpha thalassaemia is a reduced Hb synthesis due to a reduced rate (a+) or absent (a0) alpha globin synthesis Of the two alpha genes, alpha 2 usually directs a higher proportion of alpha chain synthesis and thus its deletion/mutation is more commonly implicated in thalassaemia The majority of genetic insults resulting in thalassaemia are deletions. Non-deletional mutations include: o Point mutations etc; denoted by (aTa/aa) o Mutations that give rise to a variant thalassaemic haemoglobin eg Hb constant spring (aCSa/aa) or Hb Quong Sze (aQS/aa) Unclear which is more severe out of deletion or non deletional 2. Genetic syndromes arising in alpha thalassaemia Heterozygous states o Loss of one out of four alpha genes = a+ hetero (commonest form is -a4.2/aa) Unequal recombination in meiosis giving fusion of two alpha genes on one chromosome = a+ hetero (-a3.7/aa) o Loss of both alpha genes on one chromosome = a0 hetero (--/aa) common variants in SE Asia and mediterranen, it is otherwise prevalent in Chinese, South-East Asians, Greeks, Turks, Cypriots, Sardinians Homozygous states o Loss of one alpha gene on each chromosome = a+ homo (commonest is -a4.2/-a4.2) One fusion gene each on the pair of chromosomes = a+ homo (-a3.7/-a3.7) o Loss of all four alpha genes = a0 homozygoisty (--/--) Compound heterozgosiity arises from deletions of 3 alpha genes = a+ and a0 o this is called HbH disease Genotype Name – α/ αα – α /– α – – / αα –α/–– ––/–– A+ hetero A+ homo A0 hetero Compound hetero A0 homo Phenotype α thalassaemia trait HbH disease HbBarts hydrops fetalis Clinical significance None None None Moderate Major Genetic significance None None Major Major Rather irrelevant 3. clinical syndromes arising in alpha thalassaemia Alpha-thal trait related to all heterozygous genetic states and the two a+ homozygous states harmless loss of two genes may resemble beta-thal trait; the only significance is genetic/ screening for a0 hetero to prevent conception of a child with Hb Barts DNA analysis (because all HbA, A2 and F are reduced) is necessary to diagnose a0 hetero o indices show low Hb (can be normal in A+ homo), MCV, MCH<25 HbH disease high beta:alpha chain ratio o excess beta chains form tetramers, known as HbH o HbH is ineffective at O2 transport (hyperbolic disso curve) o HbH damages RBCs = haemolytic anaemia = splenomegaly moderately severe hypochromic microcytic anaemia HbH has a ‘less worse’ anaemia than beta-thal major because the haemopoietic response to haemolysis is better and sustained, whereas it is ineffective in beta-thal because of the toxicity of a chain excess Diagnosis is made by o Indices - low Hb, MCV, MCH, MCHC o Film - golf ball cells and increased reticulocyte count o Electrophoresis o Microscopy of special HbH preparation Hb Barts & Hydrops fetalis Loss of all alpha genes means that the only Hbs that can be made are those reliant on zeta chain = Gower 1 and Portland 1; only embryonic survival is conferred o Compensation leads to production of the abnormal haemoglobins Portland 2 (z2d2) and Barts (g4) If both zeta genes are also lost then all you can make is Barts and e4 o This is seen in (--FIL/--) and (--THAI/--) o HbBarts and e4 are both non-functional, so in this case the embryo dies Unless transfusion is started in utero, this condition is incompatible with life o abortion is usually the route taken o failure to diagnose can lead to maternal death too due to HTN - need to pick up parents who both have a0 hetero 3.9 - MALARIA - read around pathology Definition A febrile illness caused by infection of red blood cells by one of four protozoal plasmodium parasites Epidemiology Incidence - 515million cases/year On the up because o More people are travelling to tropical countries o Global warming favours mosquitos o There is increased drug resistance o It is prevalent in poor countries, who are poorly equipped to deal with it, and HIV may also play a role Geography - ‘Malarial belt’ - S.America, Sub.Saharan Africa (70%), SE Asia, equatorial guinea Aetiology Causative organisms Plasmodium Falciparum o Has no life cycle outside RBCs, is becoming resistant to chloroquines and can be fatal o Higher rate of multiplication = high parasitaemia & high sequestration Sequestration confers evasion of splenic clearance Another virulence factor is antigenic variation = evading host immune response o Infects all steps of RBC lineage Plasmodium Vivax o Has a hypnozoite stage in hepatocytes, has low resistance and is rarely fatal o Preferentially invades reticulocytes Plasmodium Ovale o Also has a hypozoite stage in hepatocytes Plasmodium Malariae o Low grade persistent parasitaemia Acquisition of infection/ routes of transmission Natural - Female anopheles mosquito bite/ transplacental transmisiion o Leads to high transmission at young ages with development of immunity as adults Artificial - contamination of infected blood (transfusion, IVDU) o Leads to less solid establishment of immunity Life cycle of Plasmodium On a mosquito bite, parasitic sporozoites are injected into blood and replicate into immature schizonts within hepatocytes, which eventually burst Schizonts mature into trophozoites within RBCs o Trophozoites give rise to more schizonts = RBC rupture and cycle repeats o Trophozoites produce gametocytes which are taken up by mosquitos Gametocytes mature into sporozoites within oocytes, which eventually burst and cycle repeats Pathogenesis Cytoadherence - sequestration of parasites in deep vasculature = blood flow obstruction Rosetting is when a normal RBC adheres to an infected RBC. CD36, GAGs, complement receptor 1, Ig, fibrinogen and albumin all assist resetting Parasites prefer low oxygen so they tend to persist in post-capillary venules (low flow) Sequestration involves mainly infected cells but also some non-infected cells Excessive sequestration may be associated with organ dysfunction Plasmodium falciparum may bind to several cellular receptors (eg CD36, thrombospondin, VCAM/ICAM, selectin, GAGs) simulatenously, with pfEMP1: o Several extracellular Duffy binding-like domains (DBL 1-5) o one to two cysteine rich interdomain regions (CIDRs) between the DBLs o Transmembrane region (TM) o Submembrane/Cytoplasmic domain (Acidic terminal sequence anchored to HRP) RBC CSM changes Insertion of transport channels Modification of membrane antigens Electron dense protruberances in the erythrocyte membrane called Knobs increased deformability of non-infected RBC correlates with diseases severity o anaerobic glycolysis = acidosis o Insertion of parasite antigens on non-infected RBC Increased uptake of Ig Changes in lipid compostion Cytokines - over/unbalanced production directly causes organ damage Cytokines are produced by macrophages and T cells o Production increased in both falciparum and vivax malaria = severe disease Triggers for macrophages/ T cells to release cytokines o Soluble/exo-antigens secreted into serum. (sharp increase of TNF production) o GPI anchor - Glycosylphosphatidylinositols: anchor proteins/ polysaccharides CSM o Pigment/ Products of red cell destruction Pro-inflammatory = TNFa, IL1, 6, 8 o Harmful effects of TNFa = haemophagocytosis, hypoglycaemia, tissue damage, BM suppression, adhesion molecule production o Although some beneficial effects = phagocytosis of infected cells, pyrogenic Anti-inflammatory = IL4,5,10, TGFb o Dyserythropoiesis in chronic anaemia & Megaloblastic changes possibly due to low IL10, 12, high IFNg and macrophage dysfunction IFNg is also released by shizont recognition by T cells and leads to increased phagocytosis = persistent anaemia Mechanisms behind Anaemia Two main patterns recognised: o Acute: due to one attack in non-immune adults and young children o ‘Chronic’: repeated attacks may result in with intermittent fever Clinical features: o May be obscured by the other more dramatic complications of severe malaria o May be subtle and patient may be asymptomatic in chronic malaria o Correlates poorly with parasitaemia and reticulocyte count Three phases proposed, these often overlap o Phase 1: destruction of parasitised RBC - due to parasite burst and macrophages o Phase 2: destruction of non-parasitised RBC Non-immune mechanisms = increased deformability of noninfected RBC, reticuloendothelial hyperplasia, rarely HMS (see below), DIC Immune mechanisms = IgG, c3 dependent o Phase 3: reduced red cell production Acute malaria: Cytokine mediated suppression of erythropoiesis Reduced erythroblast iron incorporation Minor degree of dyserythropoiesis Chronic malaria - Mainly due to dyserythropoiesis BM erythroid hyperplasia suggests ineffective erythropoiesis o Increased proportion of RBCs in G2; megalobastic changes Clinical Features Symptoms/Signs/Examination Highly variable - depends on age, genetic factors, transmission and infective organism Acute malaria is a febrile illness in non-/semi-immune individuals. Watch out for severe malaria: o Severe anaemia: Hb <5.0g/dl o Cerebral malaria may give unrousable coma or seizures o Respiratory complications including ARDS and severe acidosis o Hyperparasitaemia or hyperthermia and hypoglycaemia o Renal failure Chronic malaria is typically a severe anaemia in children with Hyper reactive malarial splenomegaly (HMS) and Nephrotic syndrome if infected with P. malariae Asymptomatic malaria may occur with P. malariae Diagnosis/investigations Clinical suspicion - fever pattern, rigors, splenomegaly Blood count/film o Neutrophilia in acute malaria, monocytosis, perhaps CD4 lymphopenia o Thrombocytopenia of little clinical significance Microscopy is gold standard. Parasite species and quantitation Immunological detection of parasite antigen (Parasight™) HRP of malaria parasite Fluorescence: QBC test, DNA probes & Serology Management Conservative Fluid replacement, correction of metabolic complications (hypoglycaemia and acidosis) Ventilation, renal support if required Transfusion and exchange transfusion Medical Most antimalrial drugs are active in erythrocytic phase: (schizonticidal) o Some (primaquine) also act against tissue schizonts, hypnozoites and gametocytes Chloroquines can be used to treat Vivax, ovale and malariae o Some vivax may be resistant to chloroquine o Add Primaquine after clearance of parasitaemia (2days) to treat hepatic phase of P. ovale and P. vivax (watch out for G6PD def) IV quinine for severe falciparum infections or those unable to keep down oral medication. o Quinine For 7 days then tetracycline or Fansidar™ o Parasite clearance may take >3days o Gametocytes may persist (does not mean treatment failure) o Artemesinin and artemether derivatives also effective but still unlicensed in UK Prognosis Severe malaria has upto 20% mortality rate 3.10 - NORMAL HAEMOPOIESIS 1. Recap of developmental haemopoiesis First signs are seen in yolk sac at 3 weeks o This is Primitive haemopoiesis - there are no definitive HSCs; differentiation is believed to stem from sub-specialised subendothelial haemoangioblastic cord cells Definitive haemopoiesis begins in the AGM at 5 weeks o The evidence of HSC presence at the AGM came from repopulating ability of AGM cells (de brujn 2000) o Lack of microenvironment in AGM means it cant support HScs past 7 weeks o The fetal liver is not mature at 5 weeks o AGM would occlude if it sustained haemopoiesis Hence the transition from AGM to liver is very important for life Definitive haemopoiesis continues in the fetal liver between 7-8 weeks The BM starts to make cells from 12 weeks, but the liver and spleen are the main players in utero 2. Role of transcription factors in haemopoiesis The following transcription factors are important for initiation of haemopoiesis in the yolk sac: o LMO2, SCL/Tal1, GATA1 o They appear to cooperate; mutation in one can affect another AML1/Runx1 is an important TF for definitive haemopoiesis, but it has no role in primitive haemopoiesis Mutations in haematopoietic transcription factors are implicated in leukaemias o LMO2 mutations are implicated in T-ALL o GATA1 mutations may cause M7 AML, but only in the context of Down’s syndrome (Wechsler 2002) o Translocations in runx1 associated with AMl (8/21) and ALL (12/21) may appear in utero 3. Cells and development of different lioneages Properties of HSCs: o Undergo self renewal o Multilineage differentiation ability o Can sustain lifelong haemopiesis if transfuse into another Progenitor cells have less self-renewal capacity and are committed to a select number of lineages: o Lymphoid progenitor T, B, NK and possibly some GM cells o Myeloid everything else In order to give full differentiation of one lineage, other lineages need to be silenced o Downs syndrome demonstrates intrinsic silencing of the lymphoid lineage = myeloid overload Pathways following the common myeloid progenitor o MEP Eryhtroid is the default pathway - beginning at 3 weeks and stimulated by EPO MK starts at 5 weeks, with normal platelet counts reached by 16 weeks; TPO o GM Stimulated by GM-CSF, G-CSF and M-CSF 4. Diseases caused by stem cell abnormalities Bone Marrow failure (inc. Diamond Blackfan anaemia) Leukaemia MDS MPD 5. Clinical uses involving stem cells SCT for: o Bone marrow failure o Inherited RBC disorders - de la fuente demonstrated 10year disease free survival was 94.5% for children with haemoglobinopathy treated with AlloSCT o Haematological malignancies Repair of tissue damage (dubious evidence) Gene therapy for immunodeficieneies, inherited red cell disorders - this uses AutoSCT [see diagram] o Possibly watch out for incidence of insertional mutagenesis? French trial that gave HMGA2 activation following lentiviral transmission aiming to cure beta-thal-major had to be stopped for 2 years 3.11 - HAEMOGLOBINOPATHIES PRESENTING IN NEONATES 1. Haemoglobins present in utero/foetuses/neonates 2-4weeks (yolk sac erythroblasts) o Gower 1 (z2e2) o Gower 2 (A2E2) 4weeks (yolk sac erythroblasts/AGM/liver) o Portland (z2g2) o HbF (a2g2) 6-8weeks (liver/ spleen(3-7months)/BM) o Adult (a2b2) 2. Red cell indices in neonates Hb - 12-14 MCV - 105-120 (watch out for offsetting of a microcytosis by reticulocytes; RDW) MCH - 31-39 Platelets >150 Nucleated RBCs as a proportion of WBCs should be <20/100 3. Neonatal alpha globin disorders: A-thalassaemia and hydrops fetalis Alpha thalassaemias result from gene deletions. The size of the deletion impacts on phenotype o Large deletions involve the zeta gene (thai variant) hydrops fetalus o Smaller deletions spare the zeta gene (SEA/MED) child can be born (prematurely) without any alpha globin, because Portland (and gower1) will keep them alive Delayed blood transfusion in utero, for babies where zeta is preserved = death/neruodisability Hydrops fetalis - mechanisms of anaemia o Reduced Hb synthesis o Ineffective extramedullary haemopoiesis o Hb Barts (g4) is an unstable molecule o Hb Barts precipitates = RBC damage = haemolysis & ineffective haemopoiesis o Hb Barts has high O2 affinity so less is left for Portland Clinical features of Hb Barts/ Hydrops fetalis o Severe haemolytic anaemia with hepatosplenomegaly o Cardiac failure, oedema, pulmonary hypoplasia o Genital and limb abnornmalities o Mother may present with polyhydramnios (due to fetal anaemia) and life-threatening hypertension Look for target cells, increased eryhthroblasts, polychromasia (reticulocytes) and denatured Hb+ inclusions (on cresyl blue film) 4. Beta globin disorders in neonates Beta globin disorders present between 4-6months after birth, due to protection from HbF o Thal major presents with anaemia and failure to thrive as opposed to adult symptoms o SCD rarely presents with clinical features, there is no anaemia and blood film is normal except for HbS/HbC where they may be target cells Only HbF on HPLC could = btea thal major No HbA on HPLC could = SCA (but not ture for pre-term babies who wouldn’t have HbA anyway) 5. Gamma globin disorders in neonates Rare. They cause unexplained neonatal (<1mnth) hemolysis in the first few weeks of life that resolves as HbF dies down and HbA takes over HbPoole is a common gamma globin variant that leads to this Diagnose with HPLC/mass spectrometery 3.12 - MOLECULAR TECHNIQUES IN HAEMOGLOBINOPATHY 1. Methods in Haemoglobinpathy Diagnosis Obtaining DNA Red cells are anucleate DNA can be taken from patient’s WBCs Prenatal screening uses DNA obtained from amniocytes (amniocentesis) or chorionic villi o Samples can be taken transabdmonally or transvaginally Analysis of DNA sample Southern blotting - not used anymore; laborious and requires radioactive substances (Sanger) Sequencing (comparing DNA sequence to a normal/wild-type DNA sequence) PCR o Sample + TAQp+primers+buffer+free nucelotides o Denature at 95, extend at 72, anneal at 58; repeat GAP (primers flank mutation sites) ARMS (primers at mutation sites; 1 normal & 1 mutant) RFLP (restriction endonucleases) ASO detection system Denaturing gradient gel electrophoresis (DGGE) 2. Methods used in diagnosis of alpha thalassaemia Most common method is multiplex GAP PCR The principle is that we know alpha-thal major/ a0 hetero is down to large deletions of specified length o Hence you design primers that flank the two breakpoints of the deletion o In addition you design a primer that flanks the other side of one breakpoint o If the deletion occurs, the two flankers are brought into proximity (300-1000bp) and produce a product which is different in size to the normal, undeleted allele Hence this can detect presence of a deletion o Knowing the breakpoints/sizes of the common deletions (SEA,MED, -20.5, FIL) allows us to specifically locate the primers, and compare the products to those obtained from known alpha-thal sufferers Hence this can detect the sub-type of the deletion 3. Methods used in diagnosis of beta thalssaemia Most common method is (Sanger) sequencing on beta globin gene o The principle is that we know beta-thal major is down to mutations, so we can sequence the DNA and compare it to a normal DNA sequence to look for a difference PCR can also be used o RFLP - design restriction endonucleases that target the normal sequence at the known mutation sites For beta globin it is commonly AA 6 (S,C), 121 (D,O), 26 (E) If the mutation is present, the sequence is not cleaved by the enzyme Lack of fragmentation is evidence for a lack of mutation >1 enzyme can be used to cover all the mutation sites in one test o ARMS - design2 primers against a mutation size, with one targeting the normal sequence and one targeting a mutant sequence A control is always carried out to compare strength of signal 2 strong signals = heterozygoisyt 1 strong signal from either = homozygosity ASO detection system also used HPLC can also be used - the problem is this only tells you there is a mismatch/mutation, but not which one it is 4. Methods used in diagnosis of other red cell disorders Direct/Sanger sequencing can be applied to diagnose G6PD & PK deficiencies & diamond blackfan anaemia o The most common mutant allele in DBA is RPS19 5. Principles in next generation sequencing 2 key steps are: o Target enrichment - where you hybridise and capture Ribosomal Protein Gene DNA including exons, introns, & regulatory regions o Cluster generation - where the DNA is bridged on a flow cell and primers are added, giving a high output sequencing method Advantages o Much more useful than traditional methods when it comes to the diagnosis of disorders that are down to >1 mutation o Quick o High output - you can look at 80 genes at once o Does not require the need to know existent mutations/targeting; hence can find new mutations that would have been missed by old methods o It is expensive, but works out cost-effective because of the time it saves you o Can probably be applied to alpha & beta thalassaemia and other red cell disorders 3.13 - OVERVIEW OF RED CELL ENZYME DISORDERS 1. Overview of red cell function Transport of O2 and CO2 (and other functions achieved by Hb) Deformability, achieved via loss in intracellular contents, is key to SA/V ratio o No mitochondria = no oxidative phosphorylation = dependence on glycolysis Key Metabolic pathways o Anaerobic glycolysis = ATP o Pentose Phosphate pathway & glutathione cycle (reducing power) = resistance to oxidant damage (this is key because RBCs are particularly prone) o Pathways to remove/degrade toxic nucleotides Enzyme abnormalities result from mutations that give rise to: o A normally functioning but unstable/ prematurely deactivated protein o An abnormally functioning protein 2. General effects of red cell enzyme disorders Tend to result in a chnoric, non-spherocytic haemolytic anaemia o Extravascular haemolysis = splenomegaly Cycle; splenomegaly = pooling = worsening of anaemia o Severity varies; aplastic crises may occur and splenectomy may be required o Pigment gall stones can be common (high BR) Methaemoglobinaemia and polycythaemia as rarer phenotypes Red cells are particularly prone, and hence some unbiquitous deficiencies only affect them because: o Mutant variants are less stable in RBCs o RBCs can’t make more because they are anucleate o RBCs cant compensate via other pathways because they have no mitochondria Non-erythroid abnormalities (neurological deficit and myopathies are common) can occur with deficiencies of ubiquitous enzyme without different isoforms in different lineages Glycolytic enzymes may have other effects??? o Transcriptional/ GF regulation, stimulation of motility, apoptosis control 3. Pyruvate Kinase deficiency Definition - Congenital Deficiency of PK; A tetramer with 4 isoenzymes/isoforms; PKR is the one in RBCs; commonest glycolytic pathway deficiency Epidemiology - 1/20k in whites Pathogenesis - AR disease associated with 180 mutations; 80% of which are missense o Possible mechanism of anaemia is ATp deficiency and anti-apoptotic effects of PK giving decreased erythroblast maturation Clinical Features o Extravascular haemolysis of varying severity in childhood; spleen usually enlarged o Gallstones are common o Physiological response to chronic anaemia may lead to excess iron Investigtions o Film - normochromic anaemia, reticulocytosis, spur cells (as a mark of cellular dehydration) Spur cells can be an artefact of RBCs spending too much time in EDTA Reticulocyte count can increase 10 fold after splenectomy o Molecular methods are key to definitive diagnosis Management o Conservative - folate and iron chelation o o Medical - blood transfusion in early childhood Surgical - splenectomy for those who become transfusion dependent Gene therapy/ HSC transplant has been effective in murine models 4. Abnormalities of other glycolytic enzymes are RARE (<100cases worldwide) GP isomerase o 3rd commenst after PK and G6PD o Hetergenous presentation from mild haemolytic anaemia to death in utero o No isoenzymes = myopathies, granulocyte dysfunction, neurological deficit Hexokinase o Homozygoisty is lethal; carrier status is mild, also affects platelets Phosphofructokinase o Minimal haemolysis and mild erythrocytosis o Has multiple isoenzymes but ‘M subunit deficiency’ can cause myopathy and glycogen storage problems Triose phosphate isomerase o Severe disorder leading to death in utero or early childhood due to progressive crippling neuromuscular disease or infection o Commonest mutation is Glu104Asp Phosphoglycerate kinase - X linked disorder associated with neurological deficit and myopathy Others - aldolase, enolase, LDH 5. The Pentose phosphate and glutathione cycles & related enzyme deficiencies DEFENCE AGAINST OXIDATIVE STRESS Pentose phosphate pathway generates NADPH o Pyrimidine 5 nucelotidase catalyses the detoxification of Ump + CMP uridine + cytosine o Deficiency = nucleotide accumulation which inhibits the pentose phosphate shunt Haemolytic anaemia, unehlped by splenectomy, with basophilic stippling Glutathione cycle uses glutathione to detoxify substances o Glutathione deficiency results in chronic haemolytic anaemia, can present transiently in neonates Defects of this system = o Increased susceptibility to oxidative stress (drugs, infections, braod beans) Hb denatures and precipitates as Heinz bodies; CSM oxidisied too o This is an Intravascular haemolysis Acute haemolysis is due to irreversible damage to Hb; CSM rigidity ?Chronic haemolysis = high plasma oxidised Hb If filtered by kidneys = methaemoglobinaemia If unfiltered by kidneys = blockage = renal failure 6. Some enzyme defieincies are rare causes of Polycythaemia Bisphosphoglycerate mutase is involved in late eryhtroid differentation’ it regulates 2,3P2G level and thus O2 affinity; defiency alters affinity = polycythaemia Cytochrome b5 reductase converst metHb Hb; carriers of the deficiency get methaemoglobinaemia and homozygotes may get polycythaemia 3.14 - INDICATIONS AND CONTRAINDICATIONS OF TRANSFUSION IN RED CELL DISORDERS 1. Indications for transfusion Episodic management of: o SCA, including HbsB+thal o Bad phenotype HbH o bad phenotype beta thal intermedia Lifetime management of: o HbSB0thal o Hb constant spring o A0 thal including In utero transfusions of a0thal where gamma globin is preserved o beta thal major generally not required for enzyme and membrane disorders 2. Principles of transfusion Get right blood to right Px; identify before and after, and correct labelling of products with signature o Don’t pre label before transfusing o Insufficient clinical information will result in sample not being processed o SHOT/SAVER are two error reporting schemes; error reporting has improved Transfusion is generally warranted when benefits outweigh the risks (see below) 3. Transfusion in beta thal major Objectives Improve O2 carrying capacity Suppress ineffective haemopoiesis Maintain RBCs in circulation (not in spleen) Maintain Hb between 9 and 10.5 Iron overload & chelation 200-250mg of Fe is in each unit of transfused blood o Cannot be excreted o Utilised poorly in patients with ineffective erythropoiesis Accumulates in macrophages, overwhelms them and build in parenchyma = heart, liver and endocrine damage Chelation is usually by slow SC deferoixamine 20-60mg/kg/day over 8-24hrs Some oral iron chelators (desferiox, deferipone) are available but they have side effects (GI disturbance, neutropenia, agranulocytosis) With transfusions and compliant chelating therapy, most will live to 40-50 4. Transfusion in SCA Types of transfusion in SCA Episodic (simple transfusion) relief of crises Lifelong regimes (similar to beta thal major) aimed at maintining Hb Exchange transfusion - isovolaemic exchange Management of Clinical syndromes in SCA Aplastic crises - are usually, but not always, transient (parvovirus B19) and resolve without transfusion Splenic sequestration o This is a sudden pooling of RBCs in spleen because sickled cells get trapped in sinusoids o o Hb falls to less than 6 or drops by >3 compared to baseline Simple transfusions should be at half the expected dose to prevent viscosity, because after transfusion the spleen unloads RBCs (auto-transfusion) o Hepatic sequestration is managed in the same way Acute Stroke - STOP II trail said simple/exchange transfusion every 3-4 weeks in children with turbulent flow confirmed by Doppler, prevents first stroke; aim for HbAS/Hct <30% Acute chest syndrome - simple transfusion required within 48 hours of progressive pO2 decline; exchange transfusion for rapid deterioration Intermittent simple or exhancge transfusuions are useful for those with acute multi-organ system failure (due to infection), or patients with an isolated infection who become asymptomatically anaemic Perioperative - GA promotes IV sickling; transfusion has been shown to increase complications after surgery; but not recommended for those with Hb>7 Pulmonary HTN, Priapiasm and leg ulcers are all controversial indications of transfusion o Partial exchange transfusion in priapism can give ASPEN (Associated with SCD Priapism Exchange transfusion with Neurological complication); possibly due to ischaemia after acute Hct rise, and release of vasoactive compounds in detumesence o PDGF may be better for leg ulcers 5. Transfusion in other surgeries Eye surgery - Excessive damage to microvasculature Pregnancy - reserved for hypoxaemia, progressive persistent asymptomatic anaemia, acute chest syndrome, plenic sequestration, pre-eclampsia 6. Problems with transfusion a. Recurrent venous access becomes difficult - may require cut down procedure/ central vein access b. Transmission of infectious agents – HIV,HCV,HBV Bacteria CJD etc. c. Haemolytic transfusion reaction i. Delayed reactions occur 7-10days after transfusion, with features of haemolysis and a positive coombs test d. Immunological reactions i. In the UK most patients are black and most donors are white; poor-phenotypic compatabilty e. Transfusion associated lung injury (TRALI) f. Transfusion associated Graft v Host disease (TA-GvHD) (Rare) g. Post transfusion Purpura (PTP) h. Anaphylaxis/ Allergic /Febrile reactions i. Hyperhaemolysis syndrome i. Sevre haemolysis, reticulocytopenia, hyperbilirubinaemia, haemoglobinuria after transfusion ii. Both patient and donor cells are haemolysed iii. Coombs test usually negative 3.15 - CHALLENGES OF BLOOD TRANSFUSION IN AFRICA 1. General challenges High transfusion demand - maternal and child mortality is high Lack of resources: o Unsafe blood pool (HIV) and distribution Donors can often be lost to follow up/repeat donation o Lack of pooled resources; lack of ICT o Poor quality testing, standards and regulation Outdated equipment is a source of error Social aspects - most healthcare in Africa is private so most people can’t afford it o Hence NGOs are trying to replace hospital based services with centralised specialist ones A major challenge is developing a sustainable system 2. Sources of mortality Maternal o Pregnancy-related haemorrhage accounts for 34% maternal deaths (within 2 hours) o This is complicated by high starting rates of anaemia amongst mothers o In Africa blood can’t be given in 2 hours, antenatal screening (ABO) is unreliable and there is no records service, which would allow transfusion without X-match in an acute situation in other countires o 48% of deaths could be avoided by: Improving initial management of critical patients, and postpartum monitoring Improving availability of blood transfusions Child o 20% mortality rate below the age of 5; mostly due to malaria and lack of transfusion support 3. How a transfusion service usually works Recruit donors test and process their samples give samples to blood banks Sources of blood donors - evidence from ghana, guinea and Cameroon says volunteer are the best sources - lower Hep and HIV rates of tansmission o Voluntary Students are a major source of donors in africa Watch out for seroconversion in repeat donors o Family ?high risk infection o Commercial (paid to donate) Often poor people looking to make quick money - high risk of infection and give blood more often than is safe Solution - get rid of commercial donors and get more voluntaires. Get family members on the register so they can donate to people who aren’t their relatives Problems with recruiting donors in Africa Loss of donors/ poor donor pool (see above) o 20% of recruits are probably positive for a blood borne infection in Africa; a further 0.1% of repeat donors will acquire one o Donors can also be lost through death, lack of interest, migration Financial - Expectation of reward = decreased altruistic values = high chance of deviant behaviour Blood donation is free but there is a fee charged to patient from hospitals. Social - Fear of getting an adverse HIV sero-status report/ others knowing this Bureaucracy Lack of education Testing & processing Rapid testing is used in Africa, which is better than nothing but there is poor quality control, has a high fals positive rate, and there is the social problem of telling people they are diseased within minutes of them coming in to donate blood It can test for o HIV - EIA, p24 o HBV - HBsAg o HCV - EIA, Subtyping NAT o Syphilis - TPHA o Also malaria and other parasites, HTLV 4. Poor technology and economy are other reasons making transfusion difficult in Africa IT is needed for o reliable database about donors o traceability about who has donated what to whom o automated control of blood testing and processing o communication between transfusion centres Money is needed for o Donor recruitment, campaigning etc. o Good quality equipment for Blood testing and processing o Staff costs o It is probably more economical to centralise services in Africa rhater than put these services in every hospital There are more bodies of regulation and laws in MEDCs than Africa, outlines roles and responsibilities of blood establishments and blood banks o UK - MHRA; USA - CBER 5. Case study about Nigeria Has 6 geo-political zones, each with a regional specialist screening centre Most transfusions used to come from family and commercial donors HIv prevalence is not as high as other African countries Maternal and child mortality rates are high, as are malaria and sickle cel incidences The Abuja project 2004 did the following: o (club 25) encourages high school leavers to pledge to a healthy lifestyle and donate blood o Provided training RE knowledge, quality control, donor recruitment, safety o Introduced standards in the laboratory Lagos is a state in neigeria that does better off because it has laws restricting transfusion to licensed centres, and a paper-based records service 3.16 - AIHA & CHD 1. Classification of acquired haemolytic anaemia Immune o Warm (IgG) o Cold (igM) CHD & PNH Non-immune o Infectious o Haemophagocytic syndrome o MAHA/fragmentation syndromes o Drug induced 2. Antibodies in haemolytic anaemia & Coombs Test Auto antibodies - produced against self RBCs Alloantibodies o Self antibodies acting against foreign cells in transfusion o ‘autoantibodies’ that are secondarily acquired due to drug-induction o Placental transfer of HPLa1a? antibodies in HDN Evidence for haemolysis o Increased RBC breakdown Urine haemoglobin urobilinogen, serum bilirubin and LDH all increase Haptoglobins are all used up o Increased RBC production - reticulocytes, Erythroid hyperplasia o Morphological features SPHEROCYTES AIHA is the commonest cause of spherocytosis (small dense cells) AI damage CSM removal spherocyte; ultimately dies prematurely Polychromasia (reticulocytes) Direct antiglobulin/ coombs test - looking for and Ab already bound to RBCs o Uses antihuman complement or antihuman globulin and adds to red cell concentrate and centrifuges the mixture o Clumping is a positive result (the added antibody bridges those that are bound to RBCs) Indirect antiglobulin test - looking for presence of a Ab in a Px who has previously been sensitised (screening) o Uses recipient serum, which is removed after mixing donor RBCs, and then adds the antihuman complement/globulin as with coombs 3. Warm AIHA Definition - Autoimmune AIHA involving IgG against RBCs = Intravascular haemolytic anaemia Epidemiology - 1/75k per year; accounts for 60-70% AIHA Aetiology - 50% are idiopathic and the reminader occur in context of underlying disease eg CLL Pathogenesis - IgG binds to RBCs (targeted antigens include Rh) Clinical features - variable but often severe haemolytic anaemia, can be rapid which is an emergency (often in the elderly) or an indolent disease course Investigations - features of shortened RBC survival (see above), reticulocytes, spherocytes, and thrombocytopenia may also be present (evans syndrome) o Coombs test may show presence of IgG, C3d, or both o Elution studies also show presense of IgG Management - corticosteroids are the mainstay; 60-100mg on alternate days for three weeks achieves remission in 25-30% of patients o Splenectomy may also require maintenance low dose prednisolone o o o Rituximab (bussone 2009) achieved remisiion in 18/27; only 2 failed IVIg and plasmapheresis are other options Don’t transfuse unless theres life threatening anaemia 4. CHD Definition - Autoimmune AIHA involving IgM against RBCs = Intravascular haemolytic anaemia mediated by complement at cold temepratures. Can be acute or chronic. Epidemiology - accounts for 1./4 of AIHA Aetiology o Acute CHD can be idiopathic or secondary to Mycoplasma infection or prolymphocytic leukaemia Pathogenesis - IgM binds complement to RBCs at 4degs; the antibody dissociates on warming but complement remains and causes RBC lysis via MAC and phagocytes o Peripheries (get cold quickest) are worst affected o There are 3 types of CHD: Primary/Idiopathic Secondary to lymphoporliferative disease (PLL) Secondary to infection (mycoplasma) Clonal Ab Clonal Ab Polyclonal Ab Anti. I Anti I IgM, anti I or anti I (EBV) Clinical features - indolent chronic anaemia, exacerbated in winter; acute crises can give haemoglobinuria o Raynaud’s may also be present Investigations o Film - clumping of RBCs o Serology - IgM, complement coating of RBCs, high titre cold agglutinins and anti I antibodies (except EBV where it is anti i) Mangement - AVOID THE COLD; be wary in cold operations e.g. cardiac bypass o Chemo/rituximab may be useful for PLL patients 5. PNH Similar to CHD but mediated by IgG which means dissociation can occur at higher temperatures so haemolysis may occur at something like 20degrees (biphasic haemolysin) Usually secondary to infection (syphilis, VZV) and hence episodic and transient but severe cases may need transfusion Anti P antibodies Diagnose with Donath-lanstiener test where you take 2 samples; keep one at 0 and one at 37, warm the 0 one up to 37 and then centrifuge both; the 0 one will haemolyse because the change in temperature activates complement mediated lysis Manage by treating underlying cause 6. A B C D Other acquired haemolytic anaemias Mixed Type AIHA - both IgG and IgM; may respond to steroids IgM Warm AIHA - rare, poor prognosis Drug induced - 4 mechanisms Drug binds to RBC and attracts IgG Drug binds to Ig in serum and the complex binds to RBC Drug-Ig-RBC form and immune complex Plasma protein? Penicillin Methyl/levodopa Quinine Cephalosporins 1. 3.17 - THE SPLEEN Structure of the spleen Derived from Mesenchymal stem cell Major component of reticulo-endothelial system Can regenerate if part of it is removed Considered enlarged (USS/ CT/MRI) if >14cm by lognest axis Vasculature - Supplied by splenic artery (coeliac axis) and drained by splenic vein (portal vein) o Hence portal HTN backflow splenomegaly Histology- made of white pulp (lymphoid follicles) and red pulp composed of splenic cords (monocytes/macrophages) 2. Functions of the spleen White pulp - antibody production & a reservoir of lymphocytes Red pulp o Filtration and phagocytosis of abnormal RBCs o Removal of RBC inclusions eg howell-jolly bodies, precipitates o Pitting, malaria? o Reservoir of red cells and platelets o Synthesis of complement and F8 o Capable of haemopoiesis in BM failure 3. Causes of splenomegaly ** = massive splenomegaly = extension below umbilicus Infectious o Visceral Leishmaniasis** o Malaria (HMS**) o Bacteria (Brucella, TB) & Viruses (EBV) Haematological o Hbopathies - Thalassaemia major** (and bad intermedia), Early SCA and HbSC o MPDs - CML**, Myelofibrosis**, PV** o Lymphomas - SLVL Storage Diseases (Gauchers**) Collagen disorders Congestive (varices, portal HTN) Inflammatory - felty’s syndrome (RA) Splenomeg pooling hypersplenism o Peripheral cytopenia and anaemia (high proportion of circulating volume in spleen) 4. Splenectomy Indications o TRAUMA o ITP, hereditary spherocytosis, lymphoma, MPD, Thalassaemia Complications o Perioperative bleeding o Infections Encapsulated bacteria; pneumococcus, meningococcus, haemophilus 5 fold increase in sepsis, highest in children Give immunisations (pneumovax2) and prophylactic antibiotics o Thrombocytosis after 7-12days and hence embolism o Recurrence of initial disease (ITP) may suggest splenosis/accessory spleen (present in 15% of population); splenosis is seeding of splenic tissue onto peritoneum following rupture 5. Causes of hyposplenism Long term Splenomegaly, without splenectomy, usually turns into atrophy SCA (infarcts = fibrosis = atrophy) Long term effect of Myelofibrosis, ET, SLE, RA Coeliac, dermatitis herpetiformis Congenital absence of spleen 6. Features of hyposplenism/ splenectomy Haematological o lymphocytosis & monocytosis & reticulocytosis o Thrombocytosis, leucocytosis may also occur transiently Morphological o Persistence of things that the spleen usually removes: Howell jolly bodies (nuclear) Heinz bodies (Hb) Pappenheimer bodies (siderotic) Target cells, acanthocytes, irregularly contracted cells, nucleated RBCs, spherocytes 3.18 - TRANSPLATION IN NON-MALIGNANT DISORDERS (possibly read around this) 1. Principles of HSCT Primary objective is to achieve myeloablation and hence eradicate disease Secondary objectives are to avoid graft failure and Graft versus Host Disease This is possible by a. HLA matching i. memo - intracellular peptides are presented by class 1 & extracellular by class 2 b. Myeloablation c. Immunosuppression Stem cells can be obtained from PB, BM, Unbilical cord or via Haploidentical transplant The obtained sample can be subsequently depleted of red cells, plasma or t cells 2. Infections after HSCT Infections occur because: o Tissue damage from conditioning regimens: mouth and gut = mucositis skin o Hickman line, Neutropenia (early), Immunosuppression (late) Common infections include: o Bacterial: G-ve gut flora, Skin Staphylococcus ,Throat Streptococcus o Viral - CMV, HSV, adenovirus o Fungal - candida, aspergillus 3. Graft Failure (Host versus graft disease) Primary failure does not achieve a neutrophil count (0.5e9) within 28days Secondary failure occurs after evidence of engraftment (you achieve the count and it drops off) o Neutropenia almost always accompanied by thrombocytopenia and anaemia o Exclude the following: infections (parvovirus), drugs, GvHD, hypersplenism 4. Graft versus host disease GvHD is an immune response accentuated/ stimulated by injury from the conditioning regimen o Skin: pruritic micropapillary rash on palms, soles, or face; may become generalized o Gut: nausea, vomiting, abdominal pain, diarrhoea, bloody stools o Liver: jaundice o Lung: SOB and hypoxaemia GvHD and its treatment immunosuppression fatal infection Management options in GvHD: o Conservative - dietetic intervention with formula feeding/ TPN o Corticosteroids - topical/ systemic o Immunosuppression: e.g. tacrolimus, mycophenolate mofetil, sirolimus o Monoclonal antibodies: e.g. dacluzimab, infliximab 5. Side effects of treatment Hepatic veno-occlusive disease o In the context of HSCT, caused by total body irradiation, busulfan, cyclophosphamide o Pathology is that damaged sinusoidal endothelium sloughs = circulation block o Clinical features include painful hepatomegaly, rapid weight gain, jaundice, peripheral oedema and ascites, and possibly renal & respiratory failure if severe enough o Manage with defibrotide and fluids Increased risk of solid tumours Long term FX - infertility, pubertal failure, chronic GvHD, organ toxicity, secondary malignancy o Eggleston 2007 suggested both height and weight were impaired o Walters 2010 showed males demonstrate hypogonadotrophic hypogonadism (normal hormone levels) whereas females show primary ovarian failure (increased hormone levels); lukasa showed males demonstrate azoo/oligospermia o CNS problems - stroke/ silent infarcts (cognitive problems) Other Limitations: o Lack of donors - the eurocord study 2003 Only 18% SCD patients have a matched donor who is not affected Ethical considerations, psychological cost to the parents and financial cost o Lengthy hospital stay (2 months; followed by 4 month outpatient management) o Transplant related mortality (leukaemia module said this was upto 50%) Solution is to reduce the intensity of conditioning without compromising unwanted immune responses occuring 6. Roles of HSCT in non-malignant haematological conditions Acquired Aplastic anaemia Usually reserved for patients <40 with a matched sibling o Otherwise ATG + CsA is common Young 2010 showed matched donors have greater survival than non-identical; especially early on Stepensky and Rosenborg did work on Fludarabine as the conditioning regimen, this is now recommended if o Plt <40, Hb <9, neutrophils <1 o Persistent cytogenetic abnormalities o MDS/AML co-presence DC - alemtuzumab, cyclophosphamide, fludarabine and TBI with 200cGy is the conditioning Diamond-Blackfan 40% of patients become dependent on transfusions; the majority of the remainder who are not dependent on transfusions are dependent on steroids Several studies have shown the HSCT improves survival (75-80% 5 year survival) Matched donor is recommended for transfusion dependednt patients Unmatched donors can be used in severe circumstances eg development of aplasia/ AML/ iron overload Thalassaemia The preparatory regimen includes administration of busulfan, fludarabine and cyclophosphamide, which can eradicate the thalassemia clone, enhance immunosuppression, and facilitate sustained allogeneic engraftment Survival is influenced by the Pesaro risk classification: o Class 1 (1 risk factor) has 94% survival; 87% thal-free survival o Class 2 (1/2 risk factors) has 84% survival; 81% thal-free survival o Class 3 (3 risk factors) has 80% survival; 56% thal-free survival o Risk factors are hepatomegaly, irregular chelation and portal fibrosis HSCT associates with functional liver improvement and lower hepatic Fe conc (Mariotti 1996) SCD - Blacks do better than whites in SCA; and females do better than makles (platt 1994) 3.19 - MOLECULAR BASIS OF ALPHA THALASSAEMIA 1. Physiological abnormalities in thalssaemia On a molecular level. The body can’t detect, or rather it doesn’t downregulate what you have left (as this would worsen an already poor O2 carrying capacity), when one globin chain is thalassemic o This is why chain imbalance results from thalssaemia o This is why high HbF protects from bet-thal major in HPHF and in the neonatal period gamm globin ‘mops’ up free, cytotoxic alpha chain 2. Transcriptional environments of globin gene clusters Alpha locus is in a GC rich area = easily unwindable o Beta globin is in a GC poor area Alpha locus is in a CGP island rich area = less methylation o Beta globin is in a CGP island poor area Alpha genes are surrounded by a lot of ubiquitous/housekeeping genes = more open chromatin as it needs to be readily transcribed o Beta genes are surrounded by fewer, tissue specific genes = closed chromatin o Hence, major control of alpha genes is keeping genes off at the wrong times, whereas for beta genes it is turning genes on at the right times 3. Transcription factors in eryhtroid development In cis factors are factors produced by one chromosome that affect other genes on the same chromosome In trans factors affect genes on chromosomes different to the ones from which they originate Globin gene expression increases as erythroid cells mature Important early TFs = nuclear/ ribosomal proteins, other TFs, eg PU1 o HSCs have GATA2 and SCL targeted against HS40 o CMP has GATA1 and SCL tagrted against HS40 and local promoter regions o important) at the centre = transcription Late TFs = haem/ globin e.g GATA1 o Proerythroblasts have NFE2 poising the cell so it is ready to make globin o The thinking is that in RBCs, the polymerase complex causes DNA to loop so that all the regulatory regions come into physical proximity with the alpha genes at the centre (a2 more than a1) o Late factors may inhibit early ones to further promote differentiation (GATA1 inhibits PU1) Transcription factors bind to euchromatous regions; highly conserved regions are euchromatous 4. Epigenetic control of alpha globin transcription Heterochromatin has methylation of histones at H3K27 / K9 Euchromatin has acetylated histones and methylation of J3K4 Chromatin remodelling factors use ATP to ‘remove’ histones completely, leaving free DNA for TF binding o CRFs preferentially bind at key regulatory sites CHIP; chromatin immunoprecipitation; is a useful study for examining epigenetics o You mix DNA with formaldehde and blast it with sound waves so chromatin fragments o Then you treat some of it with anitbodies against histones, CRF, Tfs etc o You label bound and unbound chromatin with a colour each, and areas where the colours match are important and colour poor regions are not important o Lining the colours up with the DNA sequence tells you which gene it is 5. Alpha thalassaemia mutations without alpha globin deletion There are some deletions that do not involve either alpha gene but seem to overlap in a region 5’ of the a2 locus - this is probably a key regulatory element There is one syndrome where deletion of one gene (A+ hetero) presents like HbH o Not sure if the rest of this is completely true but it’s something along these lines o Here, the deletion of a1 and its stop codon leads to extended activity of a reverse mRNA 3’ of the a1 locus, which is not stopped by normal antisense mRNA (because the a1 is missing) o This leads to increased susceptibility from DNA methyltransferase o So you have deletion of one gene and methylation/downregulation of others There is a mutation that allows binding of GATA1 at the wrong site = polymerase complex centres DNA looping at the wrong site = dysfunctional mRNA produced even though both alpha genes are normal ATR-X syndrome o Presents with mild anaemia, retardation and only affects boys o ATR-X gene is on the x chromosome (in-trans factor). The inherited mutation is believed to untangle DNA to make looping and transcription easier and therefore more likely to occur at wrong sites o The idea is that the polymerase complex moves along DNA like a zip, so any site that is ‘primed’, rightly or wrongly, will get transcribed ATMDS syndrome o Acquired ATRX mutation o Unlike ATRX syndrome, it presents like HbH, it can occur in women and is not associated with mental retardation o It is believed that the differences between ATRX and ATMDS’s presentation, despite having the same mutant gene, is due to the influence of genetic changes associated with MDS 3.20 - AN ANTENATAL SERVICE FOR DIAGNOSING HAEMOGLOBINOPATHY Shit lecture; very simplistic; essay is definitely a better source of information; all that is here is the main points from what he said NHS SCT was set up in 2001 (universal screening had been in place in ‘high prevalence’ areas since the 1980s) High rpevalnce is defined as a positive test result 1.5k/10k pregnancies sct.screening.nhs.uk cpd.screening.nhs.uk/timeline otherwise everything and more is in the essay 3.21 - HAEMOGLOBINPATHY DIAGNOSIS PRACTICAL (important, read bain’s book and do online sheets) FILM Heinz bodies indicate G6PD or an unstable Hb o E.g. Hb Hammersmith Acanthocytes are spindly cells that are associated with hypospleism INDICES MCH <25 is common with a0 thal MCH <27 is common for beta thal majore ALKALINE GEL ELECTROPHORESIS Origin band CA A2/C/E S/D/G F A (Portland, barts) H Correct wording is ‘there is a band with the same mobility of…. [CSFA] Barts and H are ‘fast moving bands’ You can separate between D and G (D being a beta variant and G being an alpha variant) because G will show 3 bands Small irregular (shit looking) bands around the origin may suggest denatured Hb - unstable variant Hb Lepore moves with S ACID GEL ELECTROPHORESIS Most acid traces give a prominent F band anyway - don’t automatically think persistence of HbF/ beta thal etc HPLC Percentage of Hb in sample is demonstrated on a graph based on their retention time Furthest left is Injection artifact then, in order, is HbF, A, A2 & S o each Hb will have, in order, glycosylated (or acetylated for HbF) and degradation product peaks prior to it; these peaks always exist and may be hidden in others +VE C Origin S A/E/D/G/A2 F -VE If a variant window takes up 50% of total haemoglobin, it is probably a beta globin variant o Alpha chain variants take up 25% (4 genes) High HbA2 and HbF in beta thal trait/major (match with clinical picture and electrophoretic bands) For HbE - look for an A2 percentage which is high and roughly a third of the total haemoglobin; this is because the splicing mutation in HbE only makes the correct product 1/3 of a time For Hb Dpunjab - look for a steep window to the right of A2 o A wide based window is seen with Hb Kempsey - a variant with a high O2 affinity You need a O2 disso curve to diagnose high affinity haemoglobins Small irregular (shit looking) windows may suggest denatured Hb - unstable variant HbH shows an equal double peak to the left of HbF wheras Hb Barts is a steeper peak; looks more like hyperbilirubinaemia HbS is to the right of A2 Hb Lepore can be present if the A2 window is increased to 10-15% o Homozygoisty presents like beta-thal intermedia A delta variant called HbA2’ gives a peak that looks like S and a mildly increased A2. Add the two peaks for the true A2 value; and suspect heterogeneity with a beta-thal if the A2 total is high 3.22 - PSYCHOLOGICAL DIMENSIONS IN SCD & THALASSAEMIA 1. Initial Challenges faced by affected child Cognitive o Understanding the cause, prognosis, and complications of the disease o Need for parents to revise their expectations/visions of their childs life o Recurrent strokes can directly impair cognitive function, attention, executive function Emotional o Coming to terms with the illness o Dealing with fears/ anxieities about illness o Uptake of faith? Hoping and praying Behavioural/social o Degree of dependence of affected individual on others o Impaired social life due to treatment burden, hospital visits etc Loss of schooling and academic involvement of the child (CF cognitive challenges) o Social problems faced in concern with poor pubertal growth; body image; confidence o Disease may limit some aspects of ‘normal’ activity o Effect of disease on relationships with family members, family income etc o Affected family may become isolated 2. Issues arising in adolescence Limitations of disease burden Strain on relationships Attitudes formed against health professionals Compliance to treatment The AT&T study group 2000 said important issues are giving knowledge to the patient so they can become self-dependent. Confidence and self-efficacy increases with age and when they are confident enough, parents can relinquish care/ responsibility 3. Challenges in intervention Shifting the emphasis from hospital-based to community/ independent care Establishing home support Maintain their quality of life 4. Psychosocial Interventions that can be used Psychoeduaction - establighes groupwork, encourages coping and improves education Problem-solving CBT - encourages coping, provides emotional support and encourages self-help Neuro-education- computer programs that provide neuro-psycological assessments Expert patient programme - DH initiative that teaches patients about self-management in a chronic illness We need to incorporate psychosocial interventions into clinical management protocols 3.23 - PNH 1. Synthesis & importance of GPI GPI is a highly conserved protein bound to the outer half of the CSM Synthesis of GPI: o PIG-A adds glucosamine to IP3 on the surface of the ER o Flippase internalises the complex and mannose domains are added in the ER lumen o Phospho-ethanolamine residues are also added in ER GP anchors a number of proteins, including CD59 and other complement regulators, enzymes, adhesion molecules and co-receptors o Proteins are usually attached to phosphor-ethanolamine residues CD59 usually competes with c9 for binding sites in the C5b-7 complex; hence it prevents MAC assembly and thus complement-mediated lysis 2. Molecular basis of PNH SOMATIC MUTATION in PIG-A gene on X-chromosome o Only one mutation is needed to cause disease; women can get it o Mutations have been mapped to multiple points across the 6 exons of the PIG-A gene The mutation tends to occurs solely in HSCs o = loss of GPI synthesis = loss of GPi anchored proteins from CSM of all HSc derivatives o ‘amplified effect of one mutation’ 70% of mutations are inactivating which means there is complete GPI loss; worse phenotype o 30% of mutations are missense = partial GPI loss; better phenotype o You can have one type of mutation in one HSC; and another in another; giving two subclones CD59, CD24 and FLAER (GPI binding protein) are useful diagnostic markers The idea is that there is a fluid dynamic between the proportion of normal/mutated HSCs; and that when the balance is tipped too far; symptoms occur 3. Clinical features of PNH Intravascular haemolysis = anaemia + haemoglobinuria o It can lead to acute renal failure o Bone marrow failure, iron deficiency and folate deficiency can all further contribute to anaemia IV haemolysis = free Hb in blood = effects of depleted NO o Coaguloation related - Vasoconstiriction, thrombosis, platelet activation o SMC related - oesophageal spasms, erectile dysfunction Venous (and arterial) thromboembolism o Due to platelet activation from low CD59 levels, low uPAR, low NO and microparticle release following lysis o Not all patients are affected and it seems to be more common in the west than east o Hence PNH is probably contributory but not sufficient to cause thrombosis o Can cause budd chiari and stroke o Diagnosi with MRA/ US+doppler Bone marrow failure and cytopenias (see below) 4. Treatment of PNH Conservative - treat anaemia with iron/folate supplements MEDICAL - ECULIZUMAB o Monoclonal antibody against c5 that inhibits MAC formation without affecting upstream complement pathways o Given IV fortnightly and costs £250k/pt/year o Data from TRIUMPH trual suggests that it: Lowers LDH (a marker of RBC lysis) Lowers transfusion dependence; 49% of patients did not require a single transfusion Reduces frequency of thromboembolic events; especially in patients where >50% of granulocytes carry the mutation Improves quality of life & survival Other Treatments needed include vaccination against meningococcus/pneumococcus/haemophilus due to increased risk of infection. Prophylactic antibiotics may also be given 5. Mechanism of bone marrow failure in PNH & the role of AlloSCT There is porbably a co-exisiting, unrelated, autoimmune disease where auto-reactive T-cells target the normal HSCs and that the PIG-A mutation confers some sort of survival advantage Aims of AlloSCT are therefore to remove the abnormal HSCs and T cells and replace them with normal ones Parker 2005 said the 10year overall survival is 56% It is generally reserved for young aptients with moderate/severe bone marrow failure as they are at an increased risk of developing MDS/AML o Incidence is <5% and the mutated cells may or may not have the mutation, hence it is believed that the clones are not the reason for the development but rather the effect of the abnormal bone marrow enrivonment on other, normal cells. 3.24 - HAEMOPHAGOCYTIC LYMPHOHISTIOCYTOSIS (HLH) 1. Normal Cytotoxic T cell response to viral infection It starts with the following 2 interactions between CTL and APC: o Receptor - HLA1-TCR o Coreceptor - B7-CD28 In response to activation, T cells make perforin o Perforin and granzyme vesicles travel along actin so they get to the right target cell o Perforin makes a pore and granzyme activates caspases to mediate lysis of infected cells The activated t cell also: o Activate NK cells, which always make perforin even when they are quiescent o Activate macrophages, which then make cytokines to stimulate other T cells Regulation of T cell proliferation may be via perforin-mediated lysis of other T cells 2. Molecular basis of HLH Most mutations involve perforin loss (anywhere over 3 exons) No perforin = o Inability to lyse target cells o Uncontrolled proliferation of T cells, NK cells and macrophages o CYTOKINE STORM; Extensive production of cytokines eg TL1, Il6, TNFa o Ineffective self perpetuant inflammation and histiocytosis 3. Clinical features of HLH Infiltration of organs by Tcell/macrophages = hepatosplenomegaly, LNadenopathy, CNS involvement Haemophagocytosis by macrophages = anaemia, bone marrow failure can also occur Cytokine storm due to t cells and macrophages = coagulopathy, liver dysfunction, fever 4. Causes of HLH Inherited (rare) Familial HLH As part of another syndrome e/ge Griscelli 2, chediak higashi, X-linked lymphoproliferative Acquired (more common) Infectious - Viral, leishmania Neoplastic - EBV-lymphoma AI disease - SLE Iatrogenic Mechanisms in acquired HLh are poorly understood but viral products may inhibit exocytosis and thus release of perforin. It has a 50% mortality if untreated 5. Mutations seen in inherited HLH Familial HLH o 40% of mutations invovle the perforin gene, commonly seen in Africa (FHLH2) o Mutations in munc prevent release of a normal perforin protein from T cells, this is seen in FHLH3 (30%) and FHLH5 (10%) o 74% of Turkish patients have an STX mutation; FHLH5 Griscelli 2 o rab27 mutation means melanin can’t be delivered from melanocytes to keratinocytes = dyspigmentation (silver hair and hyperpigmentation in sun exposed areas) o Rab27 is also involved in perforin’s ability to cause pores = HLH X-linked lymphoproliferative syndrome o Only affects males (1/million) o o 50-70% of mutations involve SAP; an adapter protein in immune signalling cascades/ Other features of this syndrome are persistent EBV infections, lymphoma and increased risk of AI diseases 6. Diagnosis of HLH Molecular diagnosis o Functional assays A low cytotoxic ability of NK cells o Flow cytometry CD107a as a surrogate marker for exocytosis o Genetic analysis Test for mutations in PRF1, STX11, Munc; and look for ones that are common in certain ethnic groups 5 out of the following 8 criteria, if there is no molecular diagnosis o Fever, splenomegaly, cytopenias o High triglycerides and/or low fibrinogen o Morphological evidence of haemophgaocytosis in BM/ LNs/ spleen o No evidence of other cancer o Low or absent NK cell activity o Ferritin 500mg/l o Soluble Cd25 2400U/ml 7. Management of HLH Treat the underlying cause in acquired HLH o Amphoterecin for leishmania o Steroids and cyclosporine for AI diseases Supportive antibiotics for either inherited/acquired if there is an infection Chemo/immunotherapy for ingerited/ acquired disease to return to normal cell environment o Chemo - Eteoposide + steroids+ intrethercal methotrexate + ciclosporin o Immuno - ATG + ciclosporin (dying out because AlloSCT is better) o Monoclonal antibodies against TNFa and CD25 AlloSCT for inherited HLH only o Matched donor has much better survival than unmatched/unrelated HLH-94 treatment protocol for inherited HLH: o Start with deaily steroids, chemo, intrathecal meth, for about 8 weeks o Change to fortnightly steroids and add ciclosporin o 80% will survive until a donor becomes available 3.25 - BETA-THALASSAEMIA INTERMEDIA 1. Definition of beta thal intermedia Heteregenous condition resulting from decreased/absent globin synthesis that may be compatible with life without the need for regular transfusion o The phenotype is not always predictable based on genotype o Not all patients who are transfusion dependent to begin with need it for their whole life The reason for the heterogeneity is that the disease phenotype largely depends on the amount of globin chain imbalance o More imbalance = more ineffective erythropoiesis = symptoms You can differentiate it from Fe def anaemia on the basis of raised HbA2 NB thal cells may appear larger on blood film, but it is microcytic. The large cells are a spreading artefact that arises because cells are devoid of Hb 2. Genotypes arising in beta thal intermedia 75% of patients have 2 thalassaemic genes (homozygote) Most of these will have anaemia which is fatal without transfusion However, there are some genetic traits that prevent/ameliorate emergence of the beta-thal major phenotype, by normalising the globin chain imbalance: o The mutations inherited are mild B+ one (eg IVS6 T-C) o Compound heterozygosity o Co inheritance of alpha thalassaemia o Coinheritance of determinants that increase HbF (HPHF, delta-beta thal, Hb Lepore) 25% of patients have 1 thalassaemic gene (heterozygote) There are some genetic traits that worsen the trait/carrier phenotype, by worsening the globin chain imbalance: o Coinheritance of triplicated alpha gene (meiosis); usually mild disease o Inheritance of ‘dominant beta thalssaemia’ rare mutation that codes for a hyperunstable Hb o Coinheritance of HbE; this is probably the commonest form of thalassaemia intermedia worldwide (S.E asia) Other genetic modifiers include Alpha-Hb-stabilising protein (AHSP), which normalises the imbalance 3. Clinical features of beta thal intermedia & of standard treatment of beta thal intermedia Natural disease course Anaemia Jaundice Gallstone Bone deformities Splenomegaly Pulmonary Hypertension Leg ulcers As a result of standard treatment Haemochromatosis Psychosocial problems Inconvenience; regular hospital visits Recurrent venous access Transfuision related infection Transfusion related antibody formation (worse for intermida px than for major because they receive intermittent transfusion ata time when immune system is more mature) Splenomegaly + extramedullary haematopoiesis o Extramedullar haematopoiesis often effects paravertebral areas; pain in legs/ pain on sitting may be the first signs o Treatment options are local radio, hypertransfusion and hydroxyurea Pulmonary HTN - can occur in upto 60% of patients that are not regularly transfused as a result of free Hb and NO depletion Hemochromatosis o As a physiological response to chronic anaemia + ineffective erythropoiesis as well as due to regular transfusions o It is difficult to assess Fe loading, T2* MRI is now used to image heart and liver o Non-transferrin-bound-iron only drops for the duration of chelation; so short of 24/7 infusions, a patient will always develop iron overload Psychosocial - different for different people o Regular hospital visits = loss of education, activity, integration o Bony changes and jaundice = self consciousness o Anxiety o Sexual maturation/function problems o 81% said they would have chosen rpe natal diagnosis and abortion in hindsight 4. Other treatment methods in beta thal intermedia The OPTIMALCARE study 2010 said people who are regularly transfused and chelated have a lowed incidence of complications than those with no treatment o But you must make the decision based on patient preferences because there is no clear life expectancy difference with either avenue Oral Chelators o Deferriprone (TDS) is good for decreasing cardiomyopathy but has SFX of agranulocytosis, arthritis and GI intolerance o Deferasirox (OD) also have SFX of GI intolerance as well as renal and liver trouble o Evidence is not sure whther these are better than normal infusions Splenectomy - can riase Hb by 2g/dl and is indicated for o Hypersplenic patients; painfuk/bulky spleen o Where transfusion/chelation is not available/doable o Watch out for risk of thrombosis and sepsis after splenectomy Hydroxyurea may increase HbF levels but clinically, it is inconsistent Mini allografts are basically partial BM transplants Gene therapy may be an option for the future 3.26 - RED CELL MEMBRANE, STRUCTURE, FUNCTION & INHERITED DEFECTS 1. Basic structure of the RBC CSM It is a lipid bilayer in relationship with: o Skeletal proteins that maintain a skeleton underneath the inner layer (see below) o Trans membrane proteins studded within the membrane o (cholesterol) By weight, there are equal proportions of cholesterol and phospholipids in the CSM In terms of phospholipids, the outer membrane contains mainly PPDcholine and PPDsphingomyelin o In the inner membrane contains IP3, PPDethanolamine and PPDserine o When PPDserine is present on the outer layer; macrophages phagocytose the RBCs Maintenance of the phospholipid composition (‘dysequilibrium’) is achieved by flippase, floppase and scramblase enzymes Some important RBC membrane proteins are: o Blood group antigens (ABO, Rh) o Transport proteins Band 3 and RhAG are important for O2/CO2 transport AQP1 (water), Glut1 (glucose), Kidd (urea), ATPases and cotransporters are amongst the others o Adhesive proteins e.g. ICAM4, Lu o Signalling receptors 2. Basic functions of RBC CSM Mechanical functions o It is strong and resistant to damage o It is relatively elastic and deformable - this allows RBCs to change shape in responses to fluid stress eg. travel through small apertures eg capillaries and splenic cords o It can elongate upto 250% of its original dimensions BUT surface area is not modifiable and is kept constant; small changes in SA lead to cell lysis ‘functional’ functions o Antigen presentation (blood group) o Transport of molecules to and from the cell o Mainteneance of phospholipid ‘dysequilibirum’ and thus evasion of macrophage phagocytosis and inappropriate adhesion 3. The importance of macromolecular protein complexes for the RBC Primarily important for maintenance of structure/function relationships by allowing ‘cross talk’ between CSM proteins and intracellular skeletal proteins Ankyrin is the ‘anchor’ in the ankyrin complex; Band 3 and RhAG are connect the CSM to ankyrin vertically Protein 41R is the anchor of its omplex; Duffy antigen, Glut 1 & glycophorin connect the CSM to protein 41R vertically Anchoring proteins bind to intracellular, horizontal, skeletal proteins e.g. o Alpha and beta spectrin These are lone filamentous triple helices of repeats of 106AAs (20 repeats in alpha and 16 repeats in beta) o Actin o Adducing, dermatin, tropomyosin, tropomodulin Interactions between skeletal proteins are important in maintaining structural responses to extracellular stresses etc. 4. Structure-function relationships in the CSM RBC Geometry& maintenance of biconcave shape o The shape arises because of physical adhesion between skeletal and CSM prtoeins which prevent vesiculisation. o Chemical interacting between skeletal proteins and others maintain integrity of CSM o Cation regulation by transport proteins maintains RBC volume Cytoplasmic viscosity o Viscosity is proportional to MCH & cation concentration o High viscosity (including globin precipitates in globin synthesis disorders) hampers the cellular ability to change shape, get through small capillaries and deliver oxygen to tissues Membrane deformability o Spectrin filaments can fold/unfold rapidly; allowing the RBC to stretch o The deformability also prevents deformation-induced cell fragmentation, which would otherwise be prevalent in cases of high blood flow shear stress, physical damage in narrow capillaries etc 5. When to suspect a CSM defect Clinical Picture - (extravascular) haemolytic anaemia, jaundice, reticulocytosis, splenomegaly, gallstones Family history (most are autosomal dominant inheritance) No evidence of immune mediated haemolysis (negative coombs test) Specialised test o EMA binding assay - spherocytes don’t pick up the EMA dye o SDS PAGE electrophoresis - runnin CSM proteins eg spectrin on a gel 6. Clinical features of some hereditary RBc CSM defects Hereditary spherocytosis o AD disease, common in Caucasians, were red cells are destroyed in splenic cords o Heterogeneous presentation ranging from asymptomatic disease to anaemia require intrauterine transfusion o Thought to be due to loss of CSM SA due to mutations in proteins that are important vertical adhesive proteins in macromolecular protein complexes o Spherocytes appear small, dense, spherical and lack central pallor Hereditary elliptocytosis o AD disease; may confer protection from malaria o Also heteregenous but far fewer (10%) are asymptomatic o Thought to be due to loss of CSM SA due to mutations in proteins that are important horizontal adhesive proteins in macromolecular protein complexes o SDS PAGE electro on spectrin may show proness for spectrin to form diemrs rather than usual tetramers Hereditary stomatocytosis - AD disease where high cytoplasmic cation content gives overhydrated cells that appear as stomatocytes (increased viscosity = loss of deformability) Hereditary Ovalocytosis o Single mutation in band 3 cofners loss of deformability; common in SE Asians o Ovalocytes differ from spherocytes because they are paler and some appear as ellipses 3.27 - ACQUIRED BONE MARROW DYSFUNCTION 1. Definition of bone marrow dysfunction UNDERPRODUCTION of precursors OR production of DYSPLASTIC precursors Underproduction - aplastic anaemia, pure red cell aplasia, anaemia of chronic disease o Aplastic anaemia is hypoproliferation of all cell lineages. Acquired causes include drugs, infections and AI disease (see below) Dysplasia -MDS & dyserythropoiesis 2. Mechanisms of acquired bone marrow dysfunction Ineffective haematopoiesis due to failure of normal HSC proliferation/differentitation: o Direct infection of HSCs o Auto-immune T cells conferring cytotoxicity or producing cytokine imbalances o Abnormal bone marrow microenvironment Evidence for the role of T cells/ AI disease in bone marrow dysfunction: o Idiopathic aplastic anaemia responds to immunosuppression o Aplastic anaemia is associated with other AI cytopenias o Oligo- and clonal populations of T cells are present in many patients with aplastic anaemia, large granulocytic leukaemia and other AI cytopenias 3. Role of parvovirus This is a single stranded DNA virus which is non-enveloped It is usually spread via respiratory tract, but also via blood and placenta The protein NS1 binds to P antigen (globoside) on erythroid cells and activates caspases to cause apoptosis o It may also cause G1 cell cycle arrest; with other proteins further arresting at G2 Disease course Viral blood load starts at 5 days and peaks and about 9; antibodies follow Transient reticulocytopenia occurs for the first 5 days and there may be a subclinical anaemia Polyarthopathy may follow clearance of virus Most infections are asymptomatic but parvovirus B19 is the commonest acquired cause of pure red cell aplasia in those who have a underlying fast rate of erythropoiesis e.g.: o Patients with haemolytic anaemia, sickle cell anaemia, thalassaemia Crises are self limiting but may be fatal o Diaghnosis is made by seeing only giant erythroblasts (no reticulocytes or other precursors) in bone marrow o Recovery is followed by immunity Immunosuppressed patients eg HIV lymphoproliferative disease, iatrogenic patients may suffer from chornic pure red cell aplasia as they cannot clear the virus; give IVIg infected foetuses can die in utero Diagnosis anaemia with reticulocytopenia leads to clinical syuspicion bone marrow appearance PCR/ viral DNA hybridisation ELISA for antibodies against parvovirus (IgM during disease; IgG after immunity is acquired) 4. Role of HIV This is a retrovirus It uses gp120 to bind to CD4 cells, usually requiring coreceptors o CXCR4 in T cells and megakayrocytes o CCR5 in GM-CFU cells Advent of HAART means that haematological effects of HIV are less frequent nowadays How HIV affects the microenvironment • Effects can be mediated directly or by other microbes, which there are an increased incidence of in HIB (MAI/MAC, PCP, TB), or by an invasive lymphoma • Disrupted architecture • Disorganized architecture with increased fibrosis • Gelatinous degeneration or BM necrosis • Granulomata • Multilineage dysplasia: • Erythroblasts: nuclear irregularity and fragmentation, megaloblasts if AZT used • Myeloid cells: GMMC, nuclear fragments, abnormal chromatin pattern, apoptosis • Megakaryocytes: clustering and dysplastic forms • Increased in lymphoid cells • plasma cells especially perivascular • Increased lymphoid aggregates Effects on lymphocytes CD4 lymphopenia that correlates with viral load B cell activation; polyclonal hypergammglobulinaemia How HIV causes anaemia Dyserythropoiesis conferred by virus/ by increased susceptibility to others eg CMV, parvovirus Infiltration of BM when infections disseminate ege PCP, mycobacteria Increased risk of cancer eg NHL (DLBCL, Burkitts) and Kaposi sarcoma Malabsorption/ poor intake of B12, folate, iron Iatrogenic - AZT (megaloblastic), gancyclovir (dyserythropoiesis), chemo, antifolates Miscellaenous causes eg AIHA, TTP, DIC, Hypersplenism Treat it by treating underlying cause, dietrary supplements, EPO, transfusions How HIV causes neutropenia Dysregulated microenvironement and iatrogenic are main causes Treat underlying disease and you ould give G-CSF How HIV causes thrombocytopenia Direct megakaryocyte infection, cytokine imbalance, TTP and auto-immune (because viral proteins and GP2b3a share epitopes) are the proposed mechnaisms The phenotype is indistinguishable from ITP; anti-retrovirals, IVIg, asteroids and splenectomy are all treatment options 5. Other infections giving rise to dyserythropoiesis Malaria - can affect all three lines, commoner in chronic falciparum and vivax infections Leishmania and trypanosoma can also cause dyserythropoiesis 6. Mechanism of dyserythropoiesis in ACD Reduced production of erythrocytes: o Reduced BFUe and CFUe caused by IFN a,b and g, TNF and IL-1 o Induction of apoptosis o Reduced EPO receptors production (and thus EPO synthesis) by kidneys Dysregulation of iron metabolism o Increased production of hepcidin by the liver (stimulated by LPS & IL6): Inhibition of transferrin = less iron absorption from gut Reduced ferroportin = reduced release of iron from macrophages Stimulation of DMT1 = iron accumulates in macrophages o Increased uptake of iron by macrophages: Increased production of ferritin, DMT1 Upregulation of transferrin receptor Increased erythrophagocytosis The changes stated may not be observed in a patient who has a coexisiting iron deificnecy anaemia (where ferritin is low and thus may normalise) 3.28 - COMPLEMENT 1. Pathways in the complement system Classical pathway o Immune complexes are recognised by c1 complex o C1 complex activates c3 via c3 convertase with c4 and c2 Lectin pathway o Important for innate immunity; starts with recognition of TLRs eg MBL, ficolin, collectin 11 o Activates c3 via c4 and c2 Alternative pathway o Constitutively active; c3 auto activates itself with help of D & B proteases Terminal pathway o C3 activates c5 via c5 covertase complex o Attraction of c6-9 leads to formation of MAC; cell lysis si only directed at RBCs 2. Classical pathway C1q binds to Fc portions of IgG or IgM (but not IgG4) o Other triggers include CRP and some pathogens Binding = activation of c1q into c1r and finally c1s (serine esterase) C1s cleaves c4 into c4a and b o A fragments are released into circulation. They can recruit inflammatory cells and cleave other complement proteins o B fragments bind to CSM; c4b is an important part of both c3 and c5 convertase complexes C4b binds to c2 C1s cleaves c2 into c2a. c4b2a is known as the classical c3 convertase 3. Lectin pathway Binding of TLR activates proteases called MASPs which are analogous to c1 The rest of the pathway is the same as seen in the classical 4. Alternative pathway & terminal pathway This pathway is antibody independent and fundamental for amplification of complement response C3 binds with water and undergoes spontaneous hydrolysis. It binds to D & B protiens o D cleaves B into ba and Bb C3H20Bb is the alternative c3 covertase which rapidly cleaves c3 into c3b = auto activation C3b activates c5& initiates MAC assembly when it is bound on foreign surfaces e.g. bacteria/ cells that have poor complemetregualtory molecules e.g. RBCs o Key point in MAC assembly is polymerisation of c9 which forms a cellular pore Other functions of MAC - enzyme activation, cytokine/chemokine production, cell recovery, protein synthesis, proliferation 5. Regulation of complement Fluid/ in serum regulation o Factor H & factor I are bought into contact with C3b by sialic acid. This forms ic3b which can no longer assemble the MAC Bacteria lack sialic acid = proness to MAC mediated lysis o C1 inhibitor and c4bp inhibit the classical pathway Membrane bound regulators o CR1, DAF, MCP (c3) and CD59 (c5/MAC); all inhibit the terminal pathway Intrinsic ‘instability’ of convertase complexes Importance of regulation is in limiting damage to host tissues and preventing depletion of complement 6. Functions of complement Defence against infection o Opsonisation by c3 and c4 o Chemotaxis and leucocyte activation by c5a C5a is a powerful anaphylotoxin o Lysis by MAC (particularly encapsulated ones like Neisseria) Interface/bridge between innate and adaptive immunity o Antibody augmentation by C3 and C4 o Enhancement of memory by C3 and c4 o C3 receptors are found on B cells and APCs Waste disposal o Clearance of immune complexes, apoptotic/ dead cells; mainly by classical pathway 7. Methods of measuring complement Antigenic assays for c3 or c4 Functional assays; CH50 tests via the classical pathway and AP50 tests via the alternative pathway o Failure of both implies a terminal pathway problem 8. Role of complement is haemoatological disease Complement deficiency usually associated with recurrent infection o Classical pathway deficiency = SLE o C3 defieincy = pyogenic infections which improves with age because of mature adapative immunity o Terminal pathway deficiency = recurrent Neisseria Deficiencies of regulation Impaired fluid regulation = HUS = MAHA, renal failure, thrombocytopenia o Typical HUS is associated with cercytotoxins produced by E.Coli o Atypical HUS can be sporadic or familial and is associated with polymorphisms in one or more of Factor H, I, MCP, or high activity of B/c3 or anti-H antibody production Impaired membrane regulation = PNH = haemolgobinuria at night/early morning because low serum/urine pH increases RBC fragility o Acquired PigA mutation = loss of Cd59 = no protection from MAC o DAF deficiency increases c3 binding on RBCs but gives no cell lysis unless there is concomitant CD59 deficiency o Codeficiency of c9 may remove haemolysis o Anti c5 eculizumab is a good treatment 3.29 - GENE THERAPY IN HAEMTOLOGICAL DISORDERS 1. Requirements for gene therapy A disease where the current best treatment has failed and in which the therapeutic gene has been sequenced/cloned Cells that you can target (see below) and vectors you can use (see below) A treatment strategy (ex-vivo or in vivo??; ex-vivo better for HSCs) 2. Transient gene expression in gene therapy; causes & solutions Cause Failure to delvier to HSC Use of poorly integrating/replicating vector Immunological reactions to vector Silencing of integrated gene Solution Improve vector system or HSC prep Use a integrating/replicating vector Simplify vector, change regime, steroids Use insulator sequences 3. Target cell requierments Types of target cell - HSC/ long lived non-differntiating cell/ induced stem cell o You can induce a stem cell by reprogramming somatic cells by forcing expression of TFs like oct4, sox2, klf4 and c-myc. These are easier to grow/transfect It is difficult to do gene therapy in HSCs because there is a loss of self replication as they mature and they are difficult to isolate and transduce Solutions to problems with HSCs are o Mobilise stem cells, purify and culture o Use vectors that efficiently deliver to non-dividing cells (lentivirus) o Engineer selective advantage to transduced cells 4. Vectors The ideal vector: Have low immunogenicity Have adequate capacity for therapeutic gene Support long-term and appropriate expression Efficiently deliver gene to correct cells Be safe (fully disabled/non-mutagenic) Most vectors are viruses: Retrovirus o Removal of parts of retroviral genome e.g. gag, pol, env = no viral replication o But then you need to infect a packaging cell line to make the viral proteins needed for it to transfect the target cell Lentiviruses are a sublass of retrovirus that infect non-dividng cells better Adenovirus - host may have preexisiting immunity o DNA virus; E1, E2 and E4 domains & other coding sequences are removed o (You might need a ‘helper’ once you take out its guts) AAV - doesn’t divide without associated infection of adenovirus o 4kb small single stranded DNA virus with terminal repeats o The repeats and some promoter regions are kept in the final product Non viral vectors include cationic lipids, which form DNA-cationic lipid complexes when tranduced, and the gene gun which is only really used in plants Virus retro adeno AAV size small large small efficiency Yes - but mostly dividing Good + non dividing Good + non dividing immunogenecity low yes low mutagenesis yes No - episomal No - episomal Transient? No - provirus maintained yes yes 5. Gene therapy in SCID X linked disease associated with IL2 receptor mutation o = absent Il2 signalling (usually necessary for TCR rearrangement and Tcell maturation) o Abnormal T cells = abnormal B cells Used to be fatal within 1 year; you could live life in a bubble hoping HSCT would work French trial cured 17/20 patients but 4 developed T-ALL o The high cure rate is believed to be because the gene therapy gave infected cells a selective advantage, as well as normal function Association with ALL is because the onco-retrovirus tends to integrate near genes and its LTR region was believed to promote LMO2 6. Gene therapy in SCA/ Thalassaemia Challenges • No selective advantage for infected cells is anticipated. • High levels of b-globin expression required, erythroid specific. • Avoid oncogene inactivation. Approach efficient delivery of vector to HSC (pseudotyped lentivirus) include selectable marker into vector e.g. MDR gene include b-globin control sequences (e.g. HSs and LCRs) minimal host gene activation (e.g. SIN vectors) For diseases where no selective advantage is expected, need to: ensure high efficiency of stem cell transduction create a selective advantage: introduce selectable marker with therapeutic gene condition patient to impair proliferation of competing cells 7. Gene therapy in haemophilia - self complementary, liver specific AAV8 (see mod 1) 8. Gene therapy in cancer • Reverse mutations by expressing tumour suppressor genes/ silencing oncogenes • Virus-directed enzyme/prodrug therapy (VDEPT); bystander effect. • Stimulation of immune response to tumor; express genes for e.g.cytokines, HLA • Improve Bone Marrow Transplants; use genes that prevent GvHD 9. Gene therapy for AIDS Use genes for RNAi, Ribozymes, Interfering proteins/RNA, Intracellular antibodies Sequestration proteins (need to target multiple stages of life cycle) 10. Gene correction Safer than ‘gene replacement’ because theres less immunogenicity, less mutagenesis and no need for a virus so no chance it will get out Principle is using Zinc finger nucleases to create a DSB in order to introduce a repair construct Repair contructs are homoglous to the normal sequence and use cells own machinery to repaur DNA mutation 3.30 - DISORDERS OF GLOBIN CHAIN SYNTHESIS This was mainly a recap of stuff already covered; useful additional notes are as follows: Hydrops fetalis Large deletions that involve the zeta gene can result in hydrops fetalis. Some of these include Filipino, Thai and haawaiin variants. The SEA variant does not include zeta chain so in theory fetuses can be saved by intrauterine transfusions The anaemia in hydrops fetalis is because of inadequate Hb synthesis ( and the fact that Hb barts is pants at o2 binding/delivery), AND ineffective erythropoeises due to Hb Barts precipitates Anaemia and tissue hypoxia = o Cardiac failure o Abnormal organogenesis o Placental enlargement = pre-eclampsia and antepartum haemorrhage risks for mother Linking phenotypes to Functionally abnormal hameoglobins Sickling can be due to a variant that polymerises and/or has reduced solubility (HbS/C) High RBC could be due to a high O2 afinity Hb (Hb luton) Haemolysis could be due to an unstable Hb (Hb koln) Cyanosis could be due to a Hb that tends to oxidise (Hb M [hyde park]) Compund heterozygotes that present like SCA: Compound heterozygoisty for HbS and one of HbC/ Dpunjab/ Oarab Principle cellular changes in sickling = RIGID, ADHERENT, DEHYDRATED Compounf heterozygotes that present like thal HbBthal/HbE or lepore; delta beta thalassamia Hbconstant spring Hbathal Some other tests in diagnosis of globin chain disorders: Sickle solubility test - only identifies HbS and thus only tells you there is an SCD present; not the subtype. A positive result is turbidit/ loss of solubility Heat stability/ isopropanolol precipitation tests give a positive result (turbidity) in the presence of an unstable haemoglobin (tandem) mass spectrometry can be used to differentiate haemoglobin variants on the basis of mass and charge 3.31 - HAEMOLYTIC ANAEMIAS 1. Classifications of haemolytic anaemia (PROBABLY WORTH DOING A BETTER ONE) Defined as anaemia due to shortened red cell survival Extravascular Auto/alloimmune Hereditary spherocytosis Intravascular Malaria G6PD def Cold Autoimmune HA Mismatched (ABO) blood transfusion Drugs MAHA, TTP, PNH 2. Inherited haemolytic anaemia Can arise due to abnormalities in: CSM - proteins/ permeability Metabolism/ enzymes Hb - thalassaemia, sickling, unstable Hb variants Consequences/ clinical features Consequence Varying degree on anaemia (depending on success of EPO response/compensation) Erythroid hyperplasia, reticulocytosis Extramedullary haematopoiesis High bilirubin Free Hb in blood (if intravascular) Clinical features Pallor, SOBOE Bony pain hepatosplenomegaly Gallstones; jaundice Haemoglobinuria, NO depletion Increased folate demand can lead to folate deficiency in less developed countries Susceptibility to aplastic events following parvovirus B19 (because of low survival; remember retics would be low in this case) Chronic anaemia and reflex absorption can give Fe overload Coinheritance of Gilberts syndrome can worsen gallstone disease o Gilberts = homozygoisty for 7TA repeats = decreased BR conjugation Suspicion of inherited HA from history Young age/ congenital Episodic haemolysis may point more to something like G6PD Mode of inheritance/ family Hx Other systemic disorders (eg neuro/ myopathy trouble with enzyme deficiencies without multiple isoforms) Lab features of inherited HA Film - Anaemia, reticulocytosis, polychromasia Bloods - high BR, LDH & low (used up) haptoglobins in intravascular haemolysis Urine - haemoglobinuria (red)/ haemosiderinuria (brown) in intravascular haemolysis 3. Red cell membrane disorders Red cell cytoskeletal proteins are numbered in the order they separate on electrophoresis Herditary spherocytosis Hereditary spherocytosis is the commonest inherited HA; usually autosomal dominant It gives osmotic chsnges to RBCs - osmotic fragility test can diagnose Associated with mutations of vertical (CSM-cytoskeletal bridges) proteins; ankyrin mutations are the commenst cause of dominant HS; band 3 and spectrin mutations also seen Flow cytometry is now more frequently used in diagnosis o Eosin-5-maleimide usually binds to band 3; and band 3 content is usually reduced in hereditary spherocytosis Hereditary elliptocytosis No osmotic red cell changes Varying severity of disease Mutations in alpha spectrin mostly, alos b spectrin South east Asian Ovalocytosis Macroovalocytosis where upto half of cells may have 1 or 2 slits Heteroxzygotes may be protected against malaria; homozygotes may die because the mutation is due to deletion of band 3 4. RBC metabolic pathways G6PD is a x linked disease; may be protective against malaria o Most mutations are missense Catalyses first step in pentose phosphate pathway = generation of NADPH = maintenance of GSH Manifests as neonatal jaundice (probably because of effect of deficiency on hepatocytes) and intermittent episodes of haemolysis, triggered by oxidants eg o Drugs - anti malarials (primaquine), antibiotics (sulphonamides) o Infections, broad beans, moth balls PK deficiency is the commenst glycolytic deficiency It gives poorly deformable cells which loss K+, water, become dehydrated and are ultimately haemolysed Other metabolic pathways in RBC = 2,3 DPG shuttle, nucleotide metabolism pathwyays, GSH biosynthetic pathway (linked to pentose phosphate) and reduction of metHb by cytochrome b5 reductase 5. Investigating a haemolytic anaemia Features of haemolysis on film; heinz bodies Direct coombs (tells you if its immune or not) See ‘lab features of inherited HA’ for specific tests Thick and thin blood film for someone whose just been to a malaria endemic area; dipstick test is an alternative 6. Principles of managing haemolytic anaemia Conservative - avoiding triggers in G6PD, monitoring complications, folate Medical - transfusions for severe anaemia, immunization against HepB Surgical - cholecystectomy for recurrent gallestones. Splenectomy is good for: o Pk deficeicny, HS, severe HE and sometime in thalassaemia o Patients who are between 3-10, transfusion dependent, have poor growth and chronic Hb<8 Watch for encapsulated bacteria sepsis; give prophylactic penicillin 3.32 - G6PD DEFICIENCY 1 Structure/ Function of G6PD 59kDA dimer Binds NADP to produce NADPH (and ultimately reduced glutathione) Thus it provides protection against oxidant injury, which is important in (deformable) red cells o But, mature RBCs can make it and it’s half-life is only half that of red cell life span (old cells have less) o For example, its activity is 5x greater in reticulocytes that it is in old RBCs 2. Physiological effects of G6PD def Oxidization of Hb = Heinz bodies which attach to inner part of CSM to cause contracted cells o Is sufficient oxidation of Fe can lead to metHBaemia Oxidation of membrane proteins & lipids = cellular rigidity, dehydration Can manifest as either intra- and extravascular haemolysis 3. genetic basis of G6PD X-linked inheritance associated with mostly missense mutations o Female (heterozygotes) are effected intermediately o Mutations in exons 10/11 have the most severe deficiency and hence phenotype Sporadic mutations are rare but severe (class 1) and give a chronic haemolytic anaemia Polymorphisms are common and give intermittent episodes of haemolysis o Highest prevalence in tropical Africa, middle east, SEA and the mediterranea; selection by malaria o G6PD A- is a polymorphism that gives and unstable enzyme, mild intermittent haemolysis and occurs in Africans o G6PD Med gives severe intermittent haemolysis and is the commonest abnormality in Caucasians 4. Clinical features of G6PD Can be asymptomatic/ intermittent haemolysis or chronic haemolysis; nonspehrocytic o Severity of disease depends on premature birth, type of trigger, mutation involved o Can be fatal (G6PD Med, Chronic disease in neonatal jaundice) Intermittent episodes can be triggered by drugs, infection, acidosis, hypoxia, broad beans and lead to death of old red cells o Occurs 2-3d after usage of drug and worsens over a week; some drugs are: Dapsone Methylene blue Nitrofurantoin Primaquine Quinolones Sulphonamides o Infection is the commonest cause; watch for viruses in kids (URTi, GIT, Hep) and pneumococcus too o Occurs within 1 day of broad beans (intravascular); can also occur in babies breast feeding off a mother who has eaten broad beans; Hb drop often severe and may require transfusion Chronic (nonspherocytic) anaemia results from sporadic/class 1 mutations and is mostly extravascular and exacerbated by oxidant stress Neonatal jaundice presents possibly because of effect of deficiency on early hepatocytes, or because neonatal RBCs have high ascorbic acid, low vit E and low catalase = prone to haemolysis o Jaundice usually seen 2-3d with anaemia, reticulocytosis and may require exchange transfusion o Suspect underlying class 1 mutation if the presentation is severe 5. Diagnosis of G6PD High BR tends to occur even when there are no episodes of haemolysis o Could be unrelated to G6PD def in some cases o Could be because of impaired liver BR conjugation; coinheritance of gilberts?? Apart from clinical suspicion and film; specific G6PD tests include: o Screening, based on NADPH detection (?depletion) o Quantitative assays 6. Management of G6PD Mostly conservative - screening (especially for neonatal jaundice), education/avoidance of triggers, folate supplements for the chronic form of the disease Medical management needed for neonatal jaundice: o treat hypoxia, sepsis, acidosis promptly o phototherapy o exchange transfusion if necessary Surgical options - splenectomy may be beneficial for chronic form of the disease because it is extravascular 3.33 - HYPER REACTIVE MALARIAL SPLENOMEGALY 1. Epideimology of HRMS Age - 20-40years old Sex - generally women more than men but it varies Geography - Tends to occur in endemic malarial areas o 20% of people here have splenomegaly; massive in 4-5% o Sexual/ethnic prevalence varies by area Less common in people who have sickle trait 2. Clinical features of HRMS Many are asymptomatic; those who are present with abdo swelling/ discomfort with B symptoms Signs usually elicited are hepatosplenomegaly and signs of anaemia; fever and jaundice may occur Lab features o Film - hypersplenism signs (peripheral cytopenia), low number of malarial parasites o Marrow - hypercellular o Bloods - usually cytopenia (pooling) but you can get a polyclonal lymphocytosis sometimes o Immune - IgM, cryoglobulins, ANA, RF, thyroglobulins, immune complexes (IgM/G/C), autoantibodies, lymphocytosis 3. Pathophysiology of HRMS Mechanisms of anaemia: o splenic pooling o Reduced RBC survival/ Occasional episodes of increased haemolysis (commoner in pregnancy; coombs negative; steroids may improve) o Increased plasma volume o Increased erythropoiesis Proposed mechanism is that malaria gives chronic stimulation of B cells o Leads to IgM production and immune complex deposition in Kuppfer cells o Helper T cells are stimulated but suppressor T cells are inhibited 4. Criteria for diagnosis Major criteria o Massive splenomegaly (>10cm or below umbilicus) with no other cause (see below) o Immunity to malaria through prolonged exposure o Raised polyclonal IgM (not specific to malaria) o Improvement (40% reduction in spleen size) with malaria treatment Minor criteria o Hypersplenism o Hepatic sinusoidal T- lymphocytosis o Normal lymphocyte response to pyhtohaemoagglutinnin o PB/BM infiltiration with polyclonal lymphocytes o High antimalarial antibodiy o Family history 5. Other causes of massive splenomegaly in the tropics (differential diagnosis) Cirrhosis Portal HTN backflow of blood Infectious; kala azar & schisto Hb disorders - thalassaemia major , SS, SC Neoplastic/myeloproliferative - CML, myelofibrosis 6. Management of HRMS Medical - antimalrial therapy with daily proguanil or weekly chloroquine for atleast 12 weeks; 70% response rate Surgical - splenectomy 7. Prognosis of HRMS 50% 5 year mortality in some studies; death usually cuased by infection, anaemia or progression to lymphoma (see below) 8. HRMS as a pre-lymphomatous state Theory - High WCC is often demonstrated & lymphoma is the second commenst cause of death in HRMS patients Features of european SLVL o Epidemiology - Rare, occurs in the elderly >50, slightly males >females, western world o Features - splenomegaly, possible BM involvement & circulating Villous Lymphocytes o Diagnosis - morphology (VLs) and immunophenotypes (IgM, 20, 22, 79a, FMC7+; 23, 25-) o Prognosis - good; 80% 5 year survival African SLVL might be the same as HRMS because it presents in women who are younger and has consistent signs/ features o It is possible that a genetic mutation makes the B cells independent og malarial antigen stimulation and this results in SLVL 3.34 – RED CELL APLASIA 1. Features of Red Cell Aplasia may occur as a part of aplastic anaemia or by itself (PRCA) anaemia and reticulocytopenia are hallmarks o since red cells and platelets come from MEP; anaemia may be accompanied by thrombocytosis maturation arrest is more common that complete absence of precursors 2. Features & Associations of Acquired Pure Red Cell Aplasia rare disease; usually presents in early/middle adulthood anaemia is usually normocytic most cases are idiopathic with erythropoietic inhibitors of unknown origin May occur secondary to: o Immune diseases thymoma – 5-10% get PRCA but removal of thymus doesn’t cure PRCA AI Disease eg RA T LGL these cells are cytotoxic to RBCs o Viruses PARVOVIRUS B19 Also Hep, HIV, EBV o Pregnancy & HDN o Iatrogenic – e.g. antiepileptics, azathioprine, EPO NB this is rare noe because we use synthetic EPO with less immunogenicity 3. pathophysiology of acquired Red Cell Aplasia Immune o antibody mediated – serum inhibitors against red cells o cell mediated – as seen with T-LGL BM failure - may result in absence of progenitors rather than maturation arrest 4. Management of PRCA Conservative – stop causative drugs (including EPO) Medical o treat anaemia with transfusions and iron chelation o immunosuppression for immune mediated disease e.g. steroids, ATG, ciclosporin, Rituximab, alemtuzumab and IVIG Surgical – Splenectomy not very effective, consider BMT for non-immune disease 5. Prognosis of PRCA most people recover deaths are rarely due to aplasia; more commonly due to underlying disease e.g. AI disease, pancytopenia 6. Parvovirus B19 & PRCA This is a DNA virus viraemia lasts for a week and BM suppression occurs in this period Mostly asymptomatic, but can cause erythema in children and polyarthropathy in adults o most adults have IgG seropositivyt indicating previous asymptoimatic infection and immunity It binds to P-antigen principally of CFU-Es but P antigen is ona number of cells so it may have a wide presentation: o Transient aplasia – sudden onset anaemia which may be life threatening in people with underlying haemolytic anaemia or conditions of eryhtropoietic stress o PRCA – chronic anaemia that more commonly occurs in immunocompromised patients; diagnose with BM PCR and treat with IVIg o Hydrops Fetalis occurs because P antigen is present on placenta; so virus infects foetus occurs in 2nd trimester and may be fatal and requiring of IU transfusions o Congenital infection occurs because P antigen is present on heart (myocarditis), liver (Liver disease) and megakaryocytes (thrombocytopenia) 7. Transient erythroblastopenia of childhood TEC vs DBA in PRCA TEC may present later Normal ADA Normal HbF Recovers in 4-8 weeks DBA presents in infants <1yr High ADA High HbF does not recover TEC presents as a transient anaemia and reticulocytopenia and reduced precursors in the BM possibly caused by a virus 50% need no treatment – pbserve and only transfuse if patients develop CVS compromise 3.35 – INHERITED BONE MARROW FAILURE SYNDROMES 25% of paediatric and 10% of young adults with aplastic anaemia have underlying BM failure 1. Clinical Features of Fanconi Anaemia mean presentation is 7 years of age 30% have no abnormalities Those that are symptomatic present with BM failure and somatic abnormalities: o Skeletal deformity – no radius or thumb o skin pigmentation o short stature o horshoe kidney, ASD/VSD, hypospadias are less common Increased predisposition to cancer due to chromosome fragility o AML, Head/neck SCCs are equally common o diagnose with chromosome fragility test using diepoxybutane or mitomycin C (principle is introduce DSBs, spread metaphases and look for excessive breakage) 2. genetic basis of fanconi anaemia AR mutations of 1 of 13 FANC genes o FANCA, FANCC and FANCG are the ones most commonly mutated FANCD1 is the same as BRCA2 = higher risk of breast/ovarian Ca, brain and Wilms tumour somatic mosaicism is when FA patients don’t display chromosomal fragility because the HSC has undergone gene correction = these people present with no abnormalities but have the mutation 3. management of fanconi anaemia Conervative – avoid head/neck XR, annual BM monitoring, dentists to survey for Head/neck SCCs Medical – o HPV vaccine may protect against head/neck SCC o androgens (eg oxymetholone) support haematopoiesis Surgical – SCT o SCT doesn’t prevent future cancer; only resolves BM failure which is the main cause of detah o also consider surgery to correct somatic abnormalities, if present 4. clinical features of DBA presents as a PRCA from infancy with somatic abnormalities: o short stature present in 1/3 o hand, cardiac and renal abnormalities also common May progress to complete pancytopenia4 increased risk of developing cancer – particularly MDS, AML and osteogenic sarcomas 5. genetic basis of DBA Commonest mutations are in RPS19, RPL5, 11 = defective protein synthesis by ribosomes red cells are produced at the highest rate so this is why they are more vulnerable to protein synthesis disorders 6. management of DBA conservative – no smoking, fibre diet, no growth hormone medical – o transfusion for children less than 1 year old; 10% may enter spontaneous remission without transfusion o steroids can restore BM function in children older than 1 year; those that don’t respons rewuire chronic transfusion + chelation Surgical – SCT may resolve anaemia and reduce risk of developing myeloid neoplasm, may need additional surgery for somatic features 7. Dyskeratosis congenital Usually present in teenagers/ young adults with a typical triad: o dystrophy in nails o dyspigmentation of skin o oral leukoplakia BM failure rate is 94% by 40years old, Pulmonary fibrosis is also common 1 of the triad + BM failure + molecular evidence of mutation = diagnosis o differential diagnosis ncludes Hoyeraal-hreiadarrson syndrome (brain) and revesz syndrome (exudative retinopathy) Increased risk of cancer; solid tumours (head/neck carcinomas) more common than AML/MDS Mutations usually involve telomerase proteins – dyskerin (XL), TEKT (AD) and TERC (AR) o shorter telomeres = apoptosis/failure of chromosome replication 8. scwachman-diamond syndrome AR disease presenting in childhood with: o pancreatic exocrine insufficiency (may improve with age) o Failure to thrive o Short stature most patients have neutropenia, mechanism for this is unclear increased risk of developing aplastic anaemia, MDS by teenage years; solid tumours less common Diagnosis: o pancreatic insufficiency – low serum trypsinogen, amylase, elastase in stools, fatty pancreas on USS/CT/MRI o cytogenetics – 7-, del 7q o molecular – 95% of patients had SBDS mutation (null or hypomorphic) = dysfunctional ribosomal 60s subunit Management o conservative – fat soluble vitamins, enxyme supplements, annual BM monitoring o medical – GCSF for neutropenia o surgical – SCT (50% have hypocellular BM) 9. severe congenital neutropenia Heterogeneous prestnation at infancy spanning from neutropenia, recurrent pyogenic infections to marrow maturation arrest in granulopoiesis Most cases are AD; a variant called Kostmann SCN is inherited AR o AD – ELA2 mutations o AR – HAX133 o XL – WAS mutations o Somatic/acquired mutations may involve GCSF receptor Differntial diagnosis: o cyclic neutropenia (exclude because this rpeats every 21days; SCN doesn’t) o immune mediated disease o hypersplenism o MDS o BM examination may show maturation arrest at promyelocyte; APML management – GCSF and SCT (some evidence that SCN is a pre-leukaemic disease) 10. amegakaryocytic thrombocytopenia presents with thrombocytopenia in infants which may progress to aplastic anaemia/ AML in teens AR disease involving mutations in TPO receptor o TPO can influence the HSC which is why you can progress to aplastic anaemia o null mutations are more severe than missense mutations manage with androgens possibly, but mainly SCT 11. thrombocytopenia with absent radii congeintial BM failure, childhood haemorrhage with no radii, malformed phalanges but presence of thumbs possibly increased risk of AML, but not aplastic anaemia; TPO receptor pathway or microdeletion on choromosome 1 may be implicated platelet count usually recovers; may recover enough for orthopaedic surgery SCT rarely needed 3.36 - HAEMOGLOBIN E SYNDROMES 1. Properties to HbE Beta globin variant arising from glutamic acid lysine @ 26th AA Has a normal oxygen affinity but is slightly unstable Is produced at a reduced rate because 2/3 of the mutant transcript is nonsense and doesn’t make any protein; 1/3 makes HbE 2. Detection of HbE Electrophoresis - runs with HbA2 Heat instability test +ve Indices/film (thlassaemic/reduced rate) o Carrier - Microcytic indices MCV 75, MCH 27, Hb 12 with Hypochromic film +/- target cells; Hb E usually 1/3 of total haemoglobin produced o Homozygote - MCV 65, MCH 20, Hb 11, more abnormal film, HbE 95% of total with increased alpha chain production ratio 3. Epidemiology of HbE Sri Lanka, North India, SE asia; may offer protection from malaria o Most carriers in the UK are migrants from these areas o Compound heterozygotes in these areas may need transfusion; centres are well established in Sri Lanka; not so much in India/ Pakistan/ thaliand/ SEA Mutation may have spontaneously risen atleast 4 times 4. HbE and alpha thalassaemia Coinheritance with alpha trait reduced HbE to 20-25% Coinheritance a0/a+ compound hetero = HbH o The only globins made in this case are HbBarts and ~14% HbE Decreased chain imbalance = reduced severity of disease 5. HbE and beta thalassaemia Heterogenous presentation ranging from no symptoms to transfusion dependence o difficult to explain purely in terms of globin-chain imbalance o possible roles of Hb instability, Age, Environment o Roles of other genetic factors eg. Xmnl & epigenetic (UGT1a) polymorphisms Excess of alpha chains probably causes HbE to become unstable o RBCs cannot handle oxidative stress as well; may interact with malaria, broad beans etc o No evidence of heamolysis (but dapsone is said to cause it in 1 report) This is commonest form of sever thalassaemia in the world; Features o Dyseryhtropoietic film (not haemolytic) o Hb <10 [mean 7.6] , MCV 50-80, o No HbA is made; HbF may be raised and HbE is of the order 20-80% [mean 44%] depending on exact mutation The high HbF is useful is distinguishing this from HbE homozygoisty %HbF correlates with total Hb made; hydroxyurea may be of use 6. HbE/beta thal in the UK Presents at 4years (later) with pallor and/or acute illness and incidental diagnosis Majority of patients are Asian (Bangladeshi) migrants living in London Commonest mutation gives a svere B+ (IVS1-5;g-c); most others give a B0 70% have a XmnI polymorphism 50% become transfusion dependent at 8 years for growth failure, anaemia, acute illness therapy o 45% have splenectomies 7. Other HbE syndromes HbE/HbS - mild sickling disorder; increasingly common HbE/G6PD deficiency -no obvious phenotype HbE/HbLepore - thalassaemia intermedia, but rare HbE/HbC - symptomless; occurs in Thailand 8. Future management of HbE syndromes Improved understanding of natural history o Sri Lankan studies Noninvasive prenatal diagnosis Better prediction of phenotype from genotype Establish role of splenectomy, blood transfusion, hydroxyurea New therapeutic options o Pomalidomide, lenalidomide Management of older patients Improved iron chelation