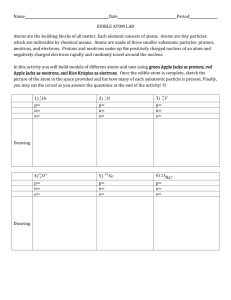
Structural, Dynamical and Electronic Properties of
advertisement

Structural, Dynamical and Electronic Properties of Transition MetalDoped Ge2Sb2Te5 Phase-Change Materials Simulated by Ab Initio Molecular Dynamics Electronic Supplementary Material 1. Repeats of Simulations The following are versions of the figures in the main text, generated from the data from the repeat simulations. Figure S1 Simulated structures of TM-doped Ge2Sb2Te5. The images above show the relaxed amorphous (top) and crystalline (bottom) structures of the (a) undoped and (b) Mn/(c) Zn-doped GST models at the end of the quench and annealing periods of the simulation. An expanded view of the local atomic geometry around the TM atoms is shown to the right of the doped models. The color-coding of the atoms is: Ge - blue, Sb - red, Te - green, Mn - purple and Zn orange. These images were produced using the VMD software (Ref. 1). - Page 2 - Bonds Per Atom 0.12 0.10 0.08 0.06 0.04 0.02 0.00 0 30 60 90 θ/ 120 150 180 Figure S2 Bond-angle distributions for the TM atoms in the amorphous (solid lines) and crystalline (dashed lines) phases. The color-coding of the lines is: Mn - purple, Zn - orange. - Page 3 - Intensity / AU (a) 80 60 40 20 0 (b) 50 100 150 Annealing Time / ps 250 60 (1) 50 MSD / Å2 200 (2) (3) 40 30 (1) 20 (3) (2) 10 0 0 50 100 150 Annealing Time / ps 200 250 Figure S3 Atomic dynamics of TM-doped Ge2Sb2Te5 during the phase transition. (a) A time-dependent Fourier analysis indicates that the undoped (black line) and Mn/Zn-doped (purple/orange lines) systems all crystallised within 150 ps of annealing. (b) The Zn mean-square displacements (MSDs) during annealing show large fluctuations, even after crystallisation (marked by an arrow on the time axis). The inset shows the geometry around the Zn atom at the three points marked on the curves. The color-coding of the atoms is as in Figure S1. - Page 4 - 0.60 Average Charge 0.50 0.40 0.30 0.20 0.10 0.00 Mn Zn Figure S4 Calculated TM-dopant atomic charges in amorphous (left, blue) and crystalline (middle, red) Ge 2Sb2Te5, compared to those in the corresponding TM telluride minerals (right, yellow). - Page 5 - 2. Transition-Metal Atom Site Preferences In the cubic Ge2Sb2Te5 (GST-225) crystal structure, transition metal (TM) dopant atoms could occupy cation or anion sites. To investigate whether there are preferences for particular sites, we carried out a series of simulations in which atoms in a crystalline model of undoped GST-225, obtained from a melt-quench-anneal molecular-dynamics simulation, were substituted with a TM atom, and the resulting model relaxed. Snapshots of the models are shown in Figure S5, and the energies of the configurations are compared in Table S1. In addition, it is possible that the TM dopant atoms could occupy interstitial sites. To test this, we took the relaxed crystalline models of the two doped systems obtained from our molecular-dynamics simulations, moved the TM atom to an interstitial site, and relaxed the resulting model. Snapshots of these models are shown in Figure S5, and the relative energies are given in the caption. Mn substitution lowers the energy of the undoped system in all cases. It is most energetically favourable for Mn to substitute at Ge/Sb (cation) sites than to replace Te at an anion site. There is a small preference for Ge sites over Sb sites in these simulations, which may be due to local distortions allowing shorter bond lengths at the former. In contrast, substitution with Zn raises the energy of the system. There appears to be a slight preference for the anion site, although there is less of a difference in energy between anion/cation substitutions than observed with Mn. It is worth noting that the relaxed models show some degree of distortion around Zn, reinforcing the idea that this atom, with its preference for tetrahedral geometry, is not well accommodated at the lattice sites in the cubic crystal structure. According to our molecular-dynamics simulations, in which all the doped systems were lower in energy than the undoped ones, Zn appears to prefer to be located near voids, where it can adopt a range of coordination geometries, which helps to explain our observations here. - Page 6 - Figure S5 Snapshots of models of crystalline Ge2Sb2Te5 in which a Ge atom (a), Sb atom (b) and Te atom (c) has been replaced by a TM atom. The top row shows the starting models, with the substituted atoms colored silver. The second row shows the relaxed Mn-substituted models, and the third row the corresponding Zn-substituted ones. The colorcoding of the atoms is: Ge - blue, Sb - red, Te - green, Mn - purple and Zn - orange. These images were produced using the VMD software (Ref. 1). - Page 7 - Figure S6 Snapshots of models of Mn (a) and Zn (b)-doped crystalline Ge2Sb2Te5 in which the TM atom has been moved to an interstitial site. The left-hand images show the starting configurations, and the right-hand ones the relaxed models. Moving the Mn atom to an interstitial site causes an energy increase of 5.69 meV per atom, while the corresponding energy change for moving Zn is 9.63 meV per atom. ∆E / meV per atom Site Mn Zn Ge -49.98 25.85 Sb -45.70 26.17 Te -36.68 21.55 Table SI Energy changes per atom on substituting Ge, Sb and Te atoms in a model of crystalline Ge2Sb2Te5 with Mn and Zn. The energy changes are calculated relative to the energy of the undoped model. - Page 8 - 3. Additional Characterisation The following are additional figures giving the results of further characterisation on the three undoped and doped systems, to supplement the information given in the main text. Unless otherwise stated, this analysis has been carried out for one of the two sets of simulations only. % Atoms (a) 100 75 50 25 0 % Atoms (b) 0 50 100 150 200 250 300 350 0 50 100 150 200 250 300 350 0 50 100 250 300 350 100 75 50 25 0 % Atoms (c) 100 75 50 25 0 150 200 Simulation Time / ps Figure S7 Dynamics of TM-doped Ge2Sb2Te5. These plots show the evolution of the percentage of atoms involved in fourfold rings (a), planes (b) and cubes (c) with time during the simulated phase-change cycle. The color coding of the lines is as follows: undoped - black, Mn-doped - purple, Zn-doped - orange. The mixing, liquid, quench and annealing periods are delineated by vertical lines. A distance cutoff of 3.5 Å, corresponding to the radius of the first coordination sphere, and an angle deviation tolerance of 20° was used to constrain these analyses; a detailed description can be found in Hegedus and Elliott (Ref. 2) (a) and Lee and Elliott (Ref. 3) (b, c). - Page 9 - (a) (b) 8.0 6.0 6.0 g(r) g(r) 4.0 4.0 2.0 2.0 0.0 0.0 0.0 1.5 3.0 4.5 r/Å 6.0 7.5 0.0 1.5 3.0 4.5 r/Å 6.0 7.5 Figure S8 Partial radial distribution functions (RDFs) centered on Mn (purple) and Zn (orange) in the amorphous (a) and crystalline (b) phases. These RDFs were computed by averaging over 4000 configurations, extending over 20 ps, at the end of the quench/annealing periods. The color-coding of the lines is as follows: Mn - purple, Zn - orange. (b) 30 30 25 25 20 20 MSD / Å2 MSD / Å2 (a) 15 10 5 15 10 5 0 0 30 35 40 45 50 Quench Time / ps 55 60 0 50 100 150 200 Annealing Time / ps 250 Figure S9 Comparison of the Mn (purple) and Zn (orange) mean-square displacements (MSDs) during (a) quenching and (b) annealing. In (b), the crystallisation times are marked by arrows. The color-coding of the lines is as in Figure S6. - Page 10 - (a) 35 30 MSD / Å2 25 20 15 10 5 0 0 100 150 200 Annealing Time / ps (c) 35 30 25 25 20 15 20 15 10 10 5 5 0 250 35 30 MSD / Å2 MSD / Å2 (b) 50 0 0 50 100 150 200 Annealing Time / ps 250 0 50 100 150 200 Annealing Time / ps 250 Figure S10 Atomic mean-square displacements (MSDs) during annealing in (a) undoped and (b) Mn-/(c) Zn-doped Ge2Sb2Te5. The color-coding of the lines is: Ge - blue, Sb - red, Te - green, Mn - purple, Zn - orange. - Page 11 - (b) 120 120 110 110 100 100 Ave. Angle Ave. Angle (a) 90 80 70 90 80 70 60 60 0 50 100 150 200 Annealing Time / ps 250 0 50 100 150 200 Annealing Time / ps 250 Figure S11 Variation in the four smallest bond angles around the Mn (a) and Zn (b) atoms during annealing. A fairly sharp reduction in the magnitude of the fluctuations occurs following crystallisation (marked by an arrow on the time axis). The bond angles considered are those for the six nearest neighbors of the TM atom. - Page 12 - (a) (b) 0.75 (c) 0.50 Average Charge Average Charge 0.50 0.25 0.00 Ge Sb Te -0.25 0.25 0.00 TM -0.50 -0.75 -0.75 (d) 0.75 Te -0.25 -0.50 0.75 0.50 0.25 0.00 Ge -0.25 Sb Te Mn Average Charge 0.50 Average Charge 0.75 0.25 0.00 Ge Sb Te Zn -0.25 -0.50 -0.50 -0.75 -0.75 Figure S12 Average Bader charges (Ref. 4) on atoms in the (a) undoped and (c) Mn-/(d) Zn-doped Ge2Sb2Te5 systems. The charges in the amorphous (blue) and crystalline (red) phases are compared. The charges on the TM atoms may be compared to those in the telluride minerals (b), MnTe (purple) and ZnTe (orange), computed from models of the unit cells of these systems. - Page 13 - Set 1 ρ / g cm-3 aGST 5.73 cGST 5.88 aGST:Mn 5.80 cGST:Mn 5.88 aGST:Zn 5.73 cGST:Zn 5.88 Set 2 Δρ ρ / g cm-3 Δρ 5.64 -2.504% 5.98 -6.025% 5.84 -1.343% 5.81 0.447% 5.52 -2.602% 5.73 -3.840% Table SII Densities of the relaxed amorphous (a-) and crystalline (c-) models of the undoped and Mn/Zn-doped systems, together with the density change on crystallisation. ∆E / meV per atom Set 1 Set 2 GST -57 -48 GST:Mn -65 -54 GST:Zn -66 -55 Table SIII Energy changes per atom on going from amorphous (a-) to crystalline(c-) phases in the undoped and Mn/Zndoped systems. The total energies compared are for relaxed configurations obtained at the end of the quench (amorphous) and annealing (crystalline) periods, and were calculated using a 2x2x2 Gamma-centred Monkhorst-Pack (Ref. 5) k-point mesh and 1.3x the plane-wave energy cutoff used during the MD runs. - Page 14 - 4. References 1 W. Humphrey, A. Dalke, and K. Schulten, Journal of Molecular Graphics 14 (1), 33 (1996). 2 J. Hegedus and S. R. Elliott, Nature Materials 7 (5), 399 (2008). 3 T. H. Lee and S. R. Elliott, Physical Review Letters 107 (14), 145702 (2011). 4 R. F. Bader, Atoms in Molecules: A Quantum Theory. (Oxford University Press, Oxford, 1990). 5 H. J. Monkhorst and J. D. Pack, Physical Review B 13 (12), 5188 (1976). - Page 15 -