Proteomics.
advertisement

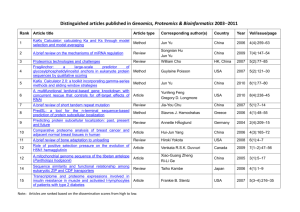
Proteomics From Wikipedia, the free encyclopedia Jump to: navigation, search For the journal Proteomics, see Proteomics (journal). Robotic preparation of MALDI mass spectrometry samples on a sample carrier. Proteomics is the large-scale study of proteins, particularly their structures and functions.[1][2] Proteins are vital parts of living organisms, as they are the main components of the physiological metabolic pathways of cells. The term "proteomics" was first coined in 1997[3] to make an analogy with genomics, the study of the genes. The word "proteome" is a blend of "protein" and "genome", and was coined by Marc Wilkins in 1994 while working on the concept as a PhD student.[4][5] The proteome is the entire complement of proteins,[4] including the modifications made to a particular set of proteins, produced by an organism or system. This will vary with time and distinct requirements, or stresses, that a cell or organism undergoes. While proteomics generally refers to the large-scale experimental analysis of proteins, it is often specifically used for protein purification and mass spectrometry. Contents 1 Complexity of the problem o 1.1 Post-translational modifications 1.1.1 Phosphorylation 1.1.2 Ubiquitination 1.1.3 Additional modifications o 1.2 Distinct proteins are made under distinct settings 2 Limitations of genomics and proteomics studies 3 Methods of studying proteins o 3.1 Identifying proteins that are post-translationally modified o 3.2 Determining the existence of proteins in complex mixtures o 3.3 Computational methods in studying protein biomarkers 4 Establishing protein–protein interactions 5 Practical applications of proteomics o 5.1 Biomarkers o 5.2 Proteogenomics o 5.3 Current research methodologies 6 See also o 6.1 Protein databases o 6.2 Research centers 7 References 8 Bibliography 9 External links [edit] Complexity of the problem After genomics and transcriptomics, proteomics is considered the next step in the study of biological systems. It is much more complicated than genomics mostly because while an organism's genome is more or less constant, the proteome differs from cell to cell and from time to time. This is because distinct genes are expressed in distinct cell types. This means that even the basic set of proteins which are produced in a cell needs to be determined. In the past this was done by mRNA analysis, but this was found not to correlate with protein content.[6][7] It is now known that mRNA is not always translated into protein,[8] and the amount of protein produced for a given amount of mRNA depends on the gene it is transcribed from and on the current physiological state of the cell. Proteomics confirms the presence of the protein and provides a direct measure of the quantity present. [edit] Post-translational modifications Not only does the translation from mRNA cause differences, but many proteins are also subjected to a wide variety of chemical modifications after translation. Many of these posttranslational modifications are critical to the protein's function. [edit] Phosphorylation One such modification is phosphorylation, which happens to many enzymes and structural proteins in the process of cell signaling. The addition of a phosphate to particular amino acids— most commonly serine and threonine[9] mediated by serine/threonine kinases, or more rarely tyrosine mediated by tyrosine kinases—causes a protein to become a target for binding or interacting with a distinct set of other proteins that recognize the phosphorylated domain. Because protein phosphorylation is one of the most-studied protein modifications, many "proteomic" efforts are geared to determining the set of phosphorylated proteins in a particular cell or tissue-type under particular circumstances. This alerts the scientist to the signaling pathways that may be active in that instance. [edit] Ubiquitination Ubiquitin is a small protein that can be affixed to certain protein substrates by enzymes called E3 ubiquitin ligases. Determining which proteins are poly-ubiquitinated can be helpful in understanding how protein pathways are regulated. This is therefore an additional legitimate "proteomic" study. Similarly, once it is determined which substrates are ubiquitinated by each ligase, determining the set of ligases expressed in a particular cell type will be helpful. [edit] Additional modifications Listing all the protein modifications that might be studied in a "Proteomics" project would require a discussion of most of biochemistry; therefore, a short list will serve here to illustrate the complexity of the problem. In addition to phosphorylation and ubiquitination, proteins can be subjected to (among others) methylation, acetylation, glycosylation, oxidation and nitrosylation. Some proteins undergo ALL of these modifications, often in time-dependent combinations, aptly illustrating the potential complexity one has to deal with when studying protein structure and function. [edit] Distinct proteins are made under distinct settings Even if one is studying a particular cell type, that cell may make different sets of proteins at different times, or under different conditions. Furthermore, as mentioned, any one protein can undergo a wide range of post-translational modifications. Therefore a "proteomics" study can become quite complex very quickly, even if the object of the study is very restricted. In more ambitious settings, such as when a biomarker for a tumor is sought – when the proteomics scientist is obliged to study sera samples from multiple cancer patients – the amount of complexity that must be dealt with is as great as in any modern biological project. [edit] Limitations of genomics and proteomics studies Proteomics typically gives us a better understanding of an organism than genomics. First, the level of transcription of a gene gives only a rough estimate of its level of expression into a protein.[10] An mRNA produced in abundance may be degraded rapidly or translated inefficiently, resulting in a small amount of protein. Second, as mentioned above many proteins experience post-translational modifications that profoundly affect their activities; for example some proteins are not active until they become phosphorylated. Methods such as phosphoproteomics and glycoproteomics are used to study post-translational modifications. Third, many transcripts give rise to more than one protein, through alternative splicing or alternative post-translational modifications. Fourth, many proteins form complexes with other proteins or RNA molecules, and only function in the presence of these other molecules. Finally, protein degradation rate plays an important role in protein content.[11] Reproducibility. Proteomics experiments conducted in one laboratory are not easily reproduced in another. For instance, Peng et al.[12] have identified 1504 yeast proteins in a proteomics experiment of which only 858 were found in a similar previous study.[13] Similarly, this latter study identified 607 proteins that were not found by Peng et al. This translates to a reproducibility of 57% (Peng vs. Washburn) to 59% (Washburn vs. Peng). [edit] Methods of studying proteins [edit] Identifying proteins that are post-translationally modified One way in which a particular protein can be studied is to develop an antibody which is specific to that modification. For example, there are antibodies which only recognize certain proteins when they are tyrosine-phosphorylated, known as phospho-specific antibodies; also, there are antibodies specific to other modifications. These can be used to determine the set of proteins that have undergone the modification of interest. For sugar modifications, such as glycosylation of proteins, certain lectins have been discovered which bind sugars. These too can be used.[citation needed] A more common way to determine post-translational modification of interest is to subject a complex mixture of proteins to electrophoresis in "two-dimensions", which simply means that the proteins are electrophoresed first in one direction, and then in another, which allows small differences in a protein to be visualized by separating a modified protein from its unmodified form. This methodology is known as "two-dimensional gel electrophoresis".[14] Recently, another approach has been developed called PROTOMAP which combines SDSPAGE with shotgun proteomics to enable detection of changes in gel-migration such as those caused by proteolysis or post translational modification.[15] [edit] Determining the existence of proteins in complex mixtures Classically, antibodies to particular proteins or to their modified forms have been used in biochemistry and cell biology studies. These are among the most common tools used by practicing biologists today. For more quantitative determinations of protein amounts, techniques such as ELISAs can be used.[citation needed] For proteomic study, more recent techniques such as matrix-assisted laser desorption/ionization (MALDI)[14] have been employed for rapid determination of proteins in particular mixtures and increasingly electrospray ionization (ESI).[citation needed] [edit] Computational methods in studying protein biomarkers Computational predictive models[16] have shown that extensive and diverse feto-maternal protein trafficking occurs during pregnancy and can be readily detected non-invasively in maternal whole blood. This computational approach circumvented a major limitation, the abundance of maternal proteins interfering with the detection of fetal proteins, to fetal proteomic analysis of maternal blood. Computational models can use fetal gene transcripts previously identified in maternal whole blood to create a comprehensive proteomic network of the term neonate. Such work shows that the fetal proteins detected in pregnant woman’s blood originate from a diverse group of tissues and organs from the developing fetus. The proteomic networks contain many biomarkers that are proxies for development and illustrate the potential clinical application of this technology as a way to monitor normal and abnormal fetal development. An information theoretic framework has also been introduced for biomarker discovery, integrating biofluid and tissue information.[17] This new approach takes advantage of functional synergy between certain biofluids and tissues with the potential for clinically significant findings not possible if tissues and biofluids were considered individually. By conceptualizing tissuebiofluid as information channels, significant biofluid proxies can be identified and then used for guided development of clinical diagnostics. Candidate biomarkers are then predicted based on information transfer criteria across the tissue-biofluid channels. Significant biofluid-tissue relationships can be used to prioritize clinical validation of biomarkers.[citation needed] [edit] Establishing protein–protein interactions Most proteins function in collaboration with other proteins, and one goal of proteomics is to identify which proteins interact. This is especially useful in determining potential partners in cell signaling cascades. Several methods are available to probe protein–protein interactions. The traditional method is yeast two-hybrid analysis. New methods include protein microarrays, immunoaffinity chromatography followed by mass spectrometry, dual polarisation interferometry, Microscale Thermophoresis and experimental methods such as phage display and computational methods [edit] Practical applications of proteomics One of the most promising developments to come from the study of human genes and proteins has been the identification of potential new drugs for the treatment of disease. This relies on genome and proteome information to identify proteins associated with a disease, which computer software can then use as targets for new drugs. For example, if a certain protein is implicated in a disease, its 3D structure provides the information to design drugs to interfere with the action of the protein. A molecule that fits the active site of an enzyme, but cannot be released by the enzyme, will inactivate the enzyme. This is the basis of new drug-discovery tools, which aim to find new drugs to inactivate proteins involved in disease. As genetic differences among individuals are found, researchers expect to use these techniques to develop personalized drugs that are more effective for the individual.[18] [edit] Biomarkers The FDA defines a biomarker as, "A characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention". Understanding the proteome, the structure and function of each protein and the complexities of protein–protein interactions will be critical for developing the most effective diagnostic techniques and disease treatments in the future. An interesting use of proteomics is using specific protein biomarkers to diagnose disease. A number of techniques allow to test for proteins produced during a particular disease, which helps to diagnose the disease quickly. Techniques include western blot, immunohistochemical staining, enzyme linked immunosorbent assay (ELISA) or mass spectrometry.[14][19] Secretomics, a subfield of proteomics that studies secreted proteins and secretion pathways using proteomic approaches, has recently emerged as an important tool for the discovery of biomarkers of disease.[20] [edit] Proteogenomics In what is now commonly referred to as proteogenomics, proteomic technologies such as mass spectrometry are used for improving gene annotations. Parallel analysis of the genome and the proteome facilitates discovery of post-translational modifications and proteolytic events,[21] especially when comparing multiple species (comparative proteogenomics).[22] [edit] Current research methodologies Fluorescence two-dimensional differential gel electrophoresis (2-D DIGE)[23] can be used to quantify variation in the 2-D DIGE process and establish statistically valid thresholds for assigning quantitative changes between samples.[24] Comparative proteomic analysis can reveal the role of proteins in complex biological systems, including reproduction. For example, treatment with the insecticide triazophos causes an increase in the content of brown planthopper (Nilaparvata lugens (Stål)) male accessory gland proteins (Acps) that can be transferred to females via mating, causing an increase in fecundity (i.e. birth rate) of females.[25] To identify changes in the types of accessory gland proteins (Acps) and reproductive proteins that mated female planthoppers received from male planthoppers, researchers conducted a comparative proteomic analysis of mated N. lugens females.[26] The results indicated that these proteins participate in the reproductive process of N. lugens adult females and males.[27] Proteome analysis of Arabidopsis peroxisomes[28] has been established as the major unbiased approach for identifying new peroxisomal proteins on a large scale.[29] There are many approaches to characterizing the human proteome, which is estimated to contain between 20,000 and 25,000 non-redundant proteins. The number of unique protein species likely increase by between 50,000 and 500,000 due to RNA splicing and proteolysis events, and when post-translational modification are also considered, the total number of unique human proteins is estimated to range in the low millions.[30][31] In addition, first promising attempts to decipher the proteome of animal tumors have recently been reported.[14] [edit] See also Activity based proteomics Bioinformatics Bottom-up proteomics Cytomics Functional genomics Genomics Immunomics Immunoproteomics Lipidomics List of biological databases List of omics topics in biology Metabolomics PEGylation Phosphoproteomics Proteogenomics Proteomic chemistry Secretomics Shotgun proteomics Top-down proteomics Systems biology Transcriptomics Yeast two-hybrid system [edit] Protein databases Cardiac Organellar Protein Atlas Knowledgebase (COPaKB) Human Protein Reference Database Model Organism Protein Expression Database (MOPED) National Center for Biotechnology Information (NCBI) Protein Data Bank (PDB) Protein Information Resource (PIR) Proteomics Identifications Database (PRIDE) Proteopedia The collaborative, 3D encyclopedia of proteins and other molecules Swiss-Prot UniProt [edit] Research centers European Bioinformatics Institute Netherlands Proteomics Centre (NPC) [edit] References 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. ^ Anderson NL, Anderson NG (1998). "Proteome and proteomics: new technologies, new concepts, and new words". Electrophoresis 19 (11): 1853–61. doi:10.1002/elps.1150191103. PMID 9740045. ^ Blackstock WP, Weir MP (1999). "Proteomics: quantitative and physical mapping of cellular proteins". Trends Biotechnol. 17 (3): 121–7. doi:10.1016/S0167-7799(98)01245-1. PMID 10189717. ^ P. James (1997). "Protein identification in the post-genome era: the rapid rise of proteomics.". Quarterly reviews of biophysics 30 (4): 279–331. doi:10.1017/S0033583597003399. PMID 9634650. ^ a b Marc R. Wilkins, Christian Pasquali, Ron D. Appel, Keli Ou, Olivier Golaz, Jean-Charles Sanchez, Jun X. Yan, Andrew. A. Gooley, Graham Hughes, Ian Humphery-Smith, Keith L. Williams & Denis F. Hochstrasser (1996). "From Proteins to Proteomes: Large Scale Protein Identification by Two-Dimensional Electrophoresis and Arnino Acid Analysis". Nature Biotechnology 14 (1): 61–65. doi:10.1038/nbt0196-61. PMID 9636313. ^ UNSW Staff Bio: Professor Marc Wilkins ^ Simon Rogers, Mark Girolami, Walter Kolch, Katrina M. Waters, Tao Liu, Brian Thrall and H. Steven Wiley (2008). "Investigating the correspondence between transcriptomic and proteomic expression profiles using coupled cluster models". Bioinformatics 24 (24): 2894–2900. doi:10.1093/bioinformatics/btn553. PMID 18974169. ^ Vikas Dhingraa, Mukta Gupta, Tracy Andacht and Zhen F. Fu (2005). "New frontiers in proteomics research: A perspective". International Journal of Pharmaceutics 299 (1–2): 1–18. doi:10.1016/j.ijpharm.2005.04.010. PMID 15979831. ^ Buckingham, Steven (May 2003). "The major world of microRNAs". http://www.nature.com/horizon/rna/background/micrornas.html. Retrieved 2009-01-14. ^ Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. (2006). "Global, in vivo, and site-specific phosphorylation dynamics in signaling networks". Cell 127 (3): 635–648. doi:10.1016/j.cell.2006.09.026. PMID 17081983. ^ Gygi, S. P.; Rochon, Y.; Franza, B. R.; Aebersold, R. (1999). "Correlation between protein and mRNA abundance in yeast". Molecular and Cellular Biology 19 (3): 1720–1730. PMC 83965. PMID 10022859. //www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=83965. edit ^ Archana Belle, Amos Tanay, Ledion Bitincka, Ron Shamir and Erin K. O’Shea (2006). "Quantification of protein half-lives in the budding yeast proteome". PNAS 103 (35): 13004–13009. doi:10.1073/pnas.0605420103. PMC 1550773. PMID 16916930. //www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1550773. ^ Peng, J.; Elias, J. E.; Thoreen, C. C.; Licklider, L. J.; Gygi, S. P. (2003). "Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: The yeast proteome". Journal of proteome research 2 (1): 43–50. PMID 12643542. edit ^ Washburn, M. P.; Wolters, D.; Yates, J. R. (2001). "Large-scale analysis of the yeast proteome by multidimensional protein identification technology". Nature Biotechnology 19 (3): 242–247. doi:10.1038/85686. PMID 11231557. edit ^ a b c d Klopfleisch R, Klose P, Weise C, Bondzio A, Multhaup G, Einspanier R, Gruber AD. (2010). "Proteome of metastatic canine mammary carcinomas: similarities to and differences from human breast cancer.". J Proteome Res 9 (12): 6380–91. doi:10.1021/pr100671c. PMID 20932060. ^ Dix MM, Simon GM, Cravatt BF (August 2008). "Global mapping of the topography and magnitude of proteolytic events in apoptosis". Cell 134 (4): 679–91. doi:10.1016/j.cell.2008.06.038. PMC 2597167. PMID 18724940. http://www.cell.com/retrieve/pii/S0092867408008258. ^ J.L. Maron , G. Alterovitz, M.F. Ramoni, K.L. Johnson, D.W. Bianchi. "High-throughput Discovery and Characterization of Fetal Protein Trafficking in the Blood of Pregnant Women", Proteomics Clinical Applications, 2009; 3(12): 1389–1396, pmid=20186258. ^ G. Alterovitz, M. Xiang, J. Liu, A. Chang, M.F. Ramoni. "System-Wide Peripheral Biomarker Discovery Using Information Theory", Pacific Symposium on Biocomputing, 2008; 231–242, "pmid=18229689". 18. ^ Vaidyanathan G. Redefining Clinical Trials: The Age of Personalized Medicine. Cell 2012;148:10791080. 19. ^ Klopfleisch R, Gruber AD. (2009). "Increased expression of BRCA2 and RAD51 in lymph node metastases of canine mammary adenocarcinomas.". Veterinary Pathology 46 (3): 416–22. doi:10.1354/vp.08-VP-0212-K-FL. PMID 19176491. 20. ^ Hathout, Yetrib (2007). "Approaches to the study of the cell secretome". Expert Review of Proteomics 4 (2): 239–48. doi:10.1586/14789450.4.2.239. PMID 17425459. 21. ^ Gupta N., Tanner S., Jaitly N., Adkins J.N., Lipton M., Edwards R., Romine M., Osterman A., Bafna V., Smith R.D., et al. Whole proteome analysis of post-translational modifications: Applications of massspectrometry for proteogenomic annotation. Genome Res. 2007;17:1362–1377. 22. ^ Gupta N., Benhamida J., Bhargava V., Goodman D., Kain E., Kerman I., Nguyen N., Ollikainen N., Rodriguez J., Wang J., et al. Comparative proteogenomics: Combining mass spectrometry and comparative genomics to analyze multiple genomes. Genome Res. 2008;18:1133–1142. 23. ^ http://onlinelibrary.wiley.com/doi/10.1002/1615-9861%28200103%291:3%3C377::AIDPROT377%3E3.0.CO;2-6/abstract 24. ^ Tonge, R., Shaw, J., Middleton, B., Rowlinson, R., Rayner, S., Young, J., Pognan, F., Hawkins, E., Currie, I. and Davison, M. (2001), Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. PROTEOMICS, 1: 377–396. doi:10.1002/16159861(200103)1:3<377::AID-PROT377>3.0.CO;2-6 25. ^ Wang, LP, Jun Shen, Lin-Quan Ge, Jin-Cai Wu, Guo-Qin Yang, Gary C. Jahn. 2010. Insecticide-induced increase in the protein content of male accessory glands and its effect on the fecundity of females in the brown planthopper, Nilaparvata lugens Stål (Hemiptera: Delphacidae). Crop Protection 29:1280-1285. 26. ^ http://pubs.acs.org/doi/pdfplus/10.1021/pr200414g 27. ^ Ge, Lin-Quan, Yao Chen , Jin-Cai Wu , and Gary C. Jahn. 2011. Proteomic analysis of insecticide triazophos-induced mating–responsive proteins of Nilaparvata lugens Stål (Hemiptera:Delphacidae). J. Proteome Res., doi:10.1021/pr200414g, Publication Date (Web): August 1, 2011. 28. ^ http://onlinelibrary.wiley.com/doi/10.1002/pmic.201000681/full 29. ^ Reumann, S. (2011), Toward a definition of the complete proteome of plant peroxisomes: Where experimental proteomics must be complemented by bioinformatics. PROTEOMICS, 11: 1764–1779. doi:10.1002/pmic.201000681 30. ^ Mathial Uhlen and Fredrik Ponten (2005). "Antibody-based Proteomics for Human Tissue Profiling". Mollecular & Cellular Proteomics 4 (4): 384–393. doi:10.1074/mcp.R500009-MCP200. 31. ^ Ole Nørregaard Jensen (2004). "Modification-specific proteomics: characterization of post-translational modifications by mass spectrometry". Current Opinion in Chemical Biology 8 (1): 33–41. doi:10.1016/j.cbpa.2003.12.009. PMID 15036154.