Electronic supplementary material - Philosophical Transactions of
advertisement

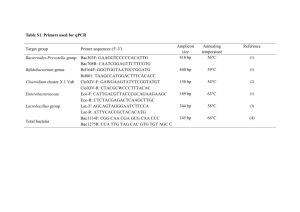
Table 1. Primers and PCR conditions used in quantitative PCR† and DGGE# with GC clamp ggcggcgcgccgcccgccccgcccccgtcgccc added to the 5‘ end*. gene 16S primer sequence 331F#† * 534r#† att acc gcg gct gct gg nirK876† aty ggc ggv cay ggc ga nirK1040 nosZ tcc tac ggg agg cag cag t qPCR primer (nM) reference 500 1 500 2 194 nirK nirS product (bp) † 5000 165 gcc tcg atc agr ttr tgg tt nirKFIaCu# atc atg gts ctg ccg cg nirKR3Cu# *gcc tcg atc agr ttg tgg tt nirS_cd3a-F#† gts aac gts aag gar acs gg nirS_R3cd-R#† 3 5000 472 4 2500 5 *gas ttc ggr tgs gtc ttg a 2500 6 nosZ2R† cak rtg cak sgc rtg gca gaa 5000 nosZ2F† cgc rac ggc aas aag gts mss gt nosZ1622RC# *cgs acc tts ttg ccs tyg cg # nosZF 410 268 7 2500 6 453 cgy tgt tcm tcg aca gcc ag To reduce heterogeneity between individual DNA extracts, each DNA sample was made from two separate aliquots of 250 mg soil from each sieved core sample used, and then pooled. For DGGE profiling duplicate PCR reactions were carried out for each of the three replicate samples. PCR amplifications for DGGE were performed in 20µl volumes with 200 µM dNTPs, 2.5 mM MgSO4, 0.2 µM Primers for 16S rRNA, nirK and nosZ and 0.4 µM for nirS primers, 3% DMSO for nosZ and 0.5µg BSA for nirS and nirK. The 16S rRNA, nirK, nirS and nosZ products were run at 60 °C on 8% acrylamide gels with 30-60%, 30-60%, 30-45% and 30-50% denaturing conditions, respectively. DGGE PCR cycling conditions - initial denaturing step 98oC (45sec) followed by 30 cycles: annealing temperature 64oC (30 sec); 8 extension temperature 72oC (30 sec); denaturing 98 oC (45sec). A final extension step at 72oC (10 min) was included. For qPCR analysis triplicate PCR reactions were carried out for each of the three replicate samples. qPCR cycling conditions - initial activation for HotStarTaq DNA polymerase at 95oC (15 min) followed by 40 cycles: annealing temperature 58oC (18 sec); extension temperature 72oC (42 sec); denaturing 95 oC (15sec). A final dissociation curve was generated to determine the quality of the applified products. References 1. Nadkarni, M.A., Martin, F.E., Jacques, N.A. & Hunter, N. 2002. Determination of bacterial load by real-time PCR using a broad range (universal) probe and primer set. Microbiology 148, 257–266. 2. Muyzer, G., Dewaal, E.C. & Uitterlinden, A.G. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reactionamplified genes coding for 16S rRNA. Appl Environ Microbiol 59, 695-700. 3. Henry, S., Baudoin, E., López-Gutiérrez, J.C., Martin-Laurent, F., Brauman, A. & Philippot, L. 2004 Quantification of denitrifying bacteria in soils by nirK gene targeted realtime PCR. J. Microbiol Methods 59, 327-335. (Erratum, 61: 289-290.) (DOI: 10.1016/j.mimet.2004.07.002; DOI: 10.1016/j.mimet.2004.12.008) 4. Hallin S. and Lindgren, P.-E. (1999) PCR detection of genes encoding nitrite reductase in denitrifing bacteria. Appl. Environ. Microbiol. 65, 1652–1657. 5. Michotey, V., Méjean, V. & Bonin, P. 2000 Comparison of methods for quantification of cytochrome cd1-denitrifying bacteria in environmental marine samples. Appl Environ Microbiol 66, 1564-1571. 6. Throbäck, I.N., Enwall, K., Jarvis, A. & Hallin, S. 2004 Reassessing PCR primers targeting nirS, nirK and nosZ genes for community surveys of denitrifying bacteria with DGGE. FEMS Microbiol Ecol 49, 401-417. (DOI: 10.1016/j.femsec.2004.04.011) 7. Henry, S., Bru, D., Stres, B., Hallet, S. and Philippot L. 2006 Quantitative detection of the nosZ gene, encoding nitrous oxide reductase, and comparison of the abundances of 16S rRNA, narG, nirK, and nosZ genes in soils. Appl Environ Microbiol 72, 5181-5189. (DOI: 10.1128/AEM.00231-06) 8. Kloos, K., Mergel, A., Rosch, C. & Bothe, H. 2001 Denitrification within the genus Azospirillum and other associative bacteria. Aust J Plant Physiol 28, 991-998.