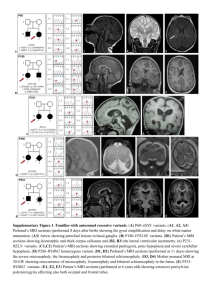
Design of the target region for gene enrichment First, a list with DCM
advertisement

Design of the target region for gene enrichment First, a list with DCM associated genes was compiled from the literature covering 84 genes that are known causes or candidate genes for DCM. To ensure a comprehensive coverage of these target genes listed in supplemental table 1, we extracted all annotated coding regions in hg19 from both, the ENSEMBL database (http://www.ensembl.org) as well as from the “University of California Santa Cruz” (UCSC) genes prediction track (http://genome.ucsc.edu), which is based on data from RefSeq, Genbank, CCDS, UniProt, Rfam and the tRNA Genes track. The resulting target region covered 496,183 bp and was used as input for eArray (Agilent Technologies, Santa Clara, California, USA) to design the custom capture-oligonucleotides for in-solution target enrichment. Manual optimization was applied to readjust capture oligonucleotides in regions with lower capture efficiency. In total, 21,641 capture probes mapping to 538,000 bp were synthesized (BED file with target region is available upon request). Gene enrichment and next-generation sequencing The amount and quality of genomic DNA samples was determined using a Qubit Fluorometer (Life Technologies; Carlsbad, CA). Then, 3 µg of genomic DNA per patient was dissolved in 135 µL 1xLow TE buffer and fragmented with the Covaris AFA system (Covaris Inc; Woburn, MA) to a size of 150 to 200 bp, as judged by DNA 1000 Bioanalyzer assay (Agilent; Waldbronn, Germany). For amplification steps during sequencing-library preparation with the SureSelectXT Target Enrichment System (Agilent; Waldbronn, Germany), the Herculase II Fusion DNA Polymerase (Agilent; Waldbronn, Germany) was used. PCR reaction was performed using a Mastercyler epGradient S (Eppendorf). The same machine was also used for hybridization of the capture oligonucleotides to the target region. The enrichment process was most efficient when hybridization was performed for 40 hours. All cleanup steps were performed with the Agencourt AMPure XP PCR purification bead system (Beckman Coulter; Pasadena, CA). The targeted DNA was captured to magnetic beads washed twice with 80% ethanol and subsequently dried at 37 degree Celsius in a PCRcycler for either 10 min or 15 min when starting volume of AMPure beads was 90 µl or 180 µl respectively. Library concentrations were measured using the Bioanalyzer DNA 1000 Chip. Six different libraries with compatible barcodes were then pooled in equal amounts and clustered with a concentration of 12 pM in one lane each of a paired end flowcell using the cBot (Illumina; San Diego, CA). Sequencing of 2x100 cycles and the 7 cycle index-run was performed on HiSeq 2000 instruments using TruSeq SBS KIT v3 chemistry (Illumina; San Diego, CA). Raw data analysis Demultiplexing of the raw sequencing reads and generation of the fastq files were done using CASAVA v.1.8.2, which was also used to calculate run metrics exemplarily displayed in supplemental table 2 from 6 consecutive sequencing runs. Mapping of the sequencing reads against human genome hg19 was done with the burrows-wheeler alignment tool (BWA v. 0.59-r16) (Li, Bioinformatics, 2009) 43. Variant calling and quality filtering of variants were performed with Genome-Analysis-Toolkit (GATK v. 1.5-21-g979a84a) (DePristo, Nature Genetics, 2011) 44 , based on alignment data where duplicate reads had previously been marked (Picard-tools 1.56) (http://picard.sourceforge.net/). For variant calling, the analysis was restricted to the target region, which was expanded by 50 bp up- and downstream of every targeted exon to allow detection of variants at splice sites. In detail, the following parameters were used: downsampling_type NONE; genotype_likelihoods_model BOTH; standard_min_confidence_threshold_for_calling 30.0; standard_min_confidence_threshold_for_emitting 30.0; p_nonref_model EXACT; heterozygosity 0.001; 2 pcr_error_rate 0.0001; genotyping_mode DISCOVERY; output_mode EMIT_VARIANTS_ONLY; min_base_quality_score 17; max_deletion_fraction 0.05; min_indel_count_for_genotyping 5; indel_heterozygosity 0.000125; indelGapContinuationPenalty 10.0; indelGapOpenPenalty 45.0; indelHaplotypeSize 80. Filtering of variants was performed using the following settings: clusterSize 3; clusterWindowSize 10; "QD < 2.0"; "MQ < 40.0"; "FS > 60.0"; "HaplotypeScore > 13.0"; "MQRankSum < -12.5"; "MQRankSumFilter"; "ReadPosRankSum < -8.0"; "QUAL < 30.0 || DP < 6 || DP > 5000 || HRun > 5"; "MQ0 >= 4 && ((MQ0 / (1.0 * DP)) > 0.1)". Insertions and deletions were filtered out using the described parameters: "QD < 2.0"; "ReadPosRankSum < -20.0; "FS > 200.0; "MQ0 >= 4 && ((MQ0 / (1.0 * DP)) > 0.1; "QUAL < 30.0 || DP < 6 || DP > 5000 || HRun > 5". Redundant variant calls between the SNP/INDEL calls were removed with the help of bedtools intersect (Quinlan AR, Bioinformatics 2010) 45. As it is known that the detection of insertions and deletions are more error prone, we manually curated all frameshift variants for miscalls. Bedtools software package was also used to calculate coverage metrics. Gene related annotation was primarily done with ANNOVAR (Wang, Nucleic Acids R. 2010 ). For some variants ANNOVAR was not able to predict a gene related effect. In these cases, SnpEff (Cingolani, Fly 2012) was used and manually curated afterwards to exclude any missannotation 46, 47 . For disease annotation, the Biobase Human Genome Mutation Database (HGMD) Genome Trax v.2013.1 was used, which we complemented by known truncating titin variants from Herman et al.48 10. Variant annotation and classification Variants were classified as benign when present in “dbSNP137common” (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/snp137Common.sql) and flagged as validated-by-frequency, which means that those variants have been found with an allelefrequency of ≥ 1% in populations. For further determination of the likelihood to being disease relevant mutations, we defined distinct categories: Category I contains all HGMD variants that are not synonymous or intronic. Category Ia contains all not common exonic variants 3 that are annotated in the HGMD database as being cardiomyopathy causing (heart muscle diseases and channelopathies) and either are non-synonymous, frameshift insertions or deletions, splice or start/stop mutations. The same definition was applied for category Ib where we additionally removed variants present in the 4300 individuals of the EuropeanAmericans cohort of the NHLBI GO Exome Sequencing Project (ESP) database (http://evs.gs.washington.edu/EVS/). Furthermore, we defined as Category II all not common truncating variants, which are either frameshift insertions/deletions, splice, or start/stop variants. Finally, all not common non-synonymous variants with prediction ‘disease’ were classified as Category III, where the prediction is based on results from the web-based tool SNPs&GO (http://snps.biofold.org/snps-and-go//snps-and-go.html) 49. PCR amplification, cloning and sequencing of individual clones For evaluation of variants with negative sanger confirmation, we first amplified the region of interest, cloned the amplicons in TOPO TA vector and sequenced individual clones. PCR for the variant LIMS1p.R19X was performed using Taq- DNA Polymerase (Qiagen, Germany) according to supplier recommendations using following primers: LIMS1-fow 5’AAGGCTGAGTCAGGTGTGCT-3’ and LIMS1-rev 5’-AGTACTTCCCAACGCTGCAT3’. Generated PCR products were subsequently extracted from a TBE agarose gel with the QIAquick Gel Kit (Qiagen, Germany) and cloned into a TOPO TA vector (Invitrogen, Carlsbad, CA, USA) following manufacturer's instructions. Plasmid DNA was isolated from randomly chosen individual bacterial colonies using GeneJET Plasmid Miniprep isolation kit (Thermo Scientific, Germany). The analysis of recombinat clones was performed using EcoRI restriction enzyme (NEB, Ipswich, MA,USA) and DNA inserts from five positive clones were sequenced by Eurofins MWG-Operon (Ebersberg Germany). By this 4 approach, we were able to successfully confirm the variant in 3 out of 5 clones (Supplemental Figure 3). 5