tpj12541-sup-0015-MethodS1-S5
advertisement

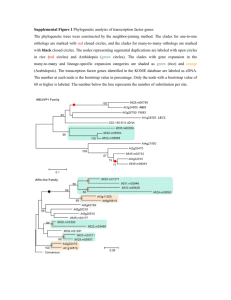
SUPPORTING INFORMATION Method S1. Construction of transgenic Arabidopsis lines Construction of transgenic Arabidopsis lines relied on the GATEWAY system (Invitrogen). Cloning cassettes from the pQE30-based expression constructs described above were amplified with primers 5’- GGGGACAAGTTTGTACAAAAAAGCAGGCTCTATGAGAGGATCGCATCACC-3’ and 5’- GGGACCACTTTGTACAAGAAAGCTGGGTACAGGAGTCCAAGCTCA-3’, which introduced recombination sites attP1 and attP2, allowing for recombination into the pDONR221 vector (Invitrogen, Carlsbad, CA, USA) via Gateway Clonase II BP. The fidelity of the entry clones was checked by sequencing using standard M13 forward and reverse primers. The destination clone was created by recombination of linearized pDONR221 plasmid into pGWB2 or pGWB6 (Tsuyoshi Nakagawa, Research Institute of Molecular Genetics, Shimane University, Matsue 690, Japan) with the use of Gateway Clonase II LR. Destination vectors placed the UGTs under the control of CaMV 35S promoter and provided kanamycin resistance. While pGWB2 vector was designed to produce the His-tagged construct, pGWB6 also provided a C-terminal GFP protein tag. The destination vectors were transformed into Agrobacterium tumefaciens GV3101 and used to transform heterozygous ugt74b1-2 plants, allowing isolation of each overexpression insertion event in both wild-type and the ugt74b1-2 backgrounds. Plants were transformed by the floral dip method (Clough and Bent, 1998). Seeds collected from primary transformants were re-screened in the T2 generation to identify lines harboring single insertions, and progeny of individual T2 plants were screened on both kanamycin and Basta to determine the genotype (homozygous or heterozygous) of the T2 parent with respect to the overexpression construct (KanR) and the ugt74b1-2 insertion (Bar). 1 Method S2. Plant growth conditions Soil grown plants were planted in Pro-Mix ‘BX’ (Premier Horticulture, Red Hill, PA, USA) at 22°C and 70% relative humidity under illumination with fluorescent and incandescent light (16 h per day) at a photon fluence rate of approximately 120 mol m-2 sec-1. Plants were watered 2-3 times per week and fertilized with macronutrients (7.5 mM N, 1.7 mM P, 3 mM K, 3 mM Ca, and 1.2 mM Mg). For plant growth in sterile conditions, seeds were surfacesterilized for 10 min in 1.5% (w/v) sodium hypochlorite and placed on solid medium containing 8 g l-1 phytagar, 30 g l-1 sucrose, 2.15 g l-1 (0.5 ×) Murashige-Skoog (MS) salts, pH 5.6, and 1 × MS vitamin mix (Sigma, St. Louis, MO, USA). After 2-3 days of stratification at 4oC, plants were grown at 24oC at a photon fluence rate of approximately 160 mol m-2 sec-1 for 18 h (long days) or 8 h (short days) per day. For selection of transgenic plants, the agar medium was supplemented with 25 μg ml-1 kanamycin (Sigma), or with 5.25 g ml-1 sulfadiazine (Sigma), or seedlings grown in soil were sprayed with 1% (v/v) Basta herbicide (Finale, Farnam Companies, Phonix, AZ, USA). Method S3. Analysis of endogenous hormone contents Fresh plant material (10-80 mg) was homogenized with 5 ml methanol and 50 ng each of (D4)ACC (1-aminocyclopropane-1-carboxylic acid), (2H5)JA (jasmonic acid), (2H5)OPDA (12-oxo-phytodienoic acid), (-)JA-(2H3)Leu (jasmonate-isoleucine), (D6)ABA (abscisic acid), (13C6)IAA (indolyl-3-acetic acid) and [13C6, 15N1]IBA (indolyl-3-butyric acid) were added as internal standards. Extract preparation, derivatization and GC-MS analysis (Polaris Q, Thermo-Finnigan, Bremen, Germany) were performed as previously described (Grubb et al., 2004; Miersch et al., 2008; Sreenivasulu at al., 2010). 2 Method S4. Phylogenetic analysis of the UGT74 clade A search for proteins related to the UGT74 clade employed blastp searches at the NCBI website (http://blast.ncbi.nlm.nih.gov/Blast.cgi) with default parameters, except the organism field was restricted to green plants (taxid:33090). The following proteins were used as query sequences: UGT74B1, UGT74C1, UGT74D1, UGT74E1, UGT74F1 and UGT75C1. The latter was included because a previous phylogenetic analysis of Arabidopsis Family 1 UGTs indicated that it is closely related to, but outside of, the UGT74 clade (Li et al., 2001). An “E value” of 1 x 10-78 was chosen as the cutoff; this was the least stringent value for which every query recovered at least one sequence each from the genomes of Zea mays, Oryza sativa and Sorghum bicolor. The recovered gene lists were combined, duplicates discarded and a total of 269 records retrieved. One sequence we believe to be misannotated (UGT74E1) was corrected in both A. thaliana and A. lyrata. An initial tree was constructed, and the position of UGT75C1 was used to make a final determination of which sequences to include in further analyses. Finally, UGT74 homologs from Carica papaya were retrieved from PLAZA (http://bioinformatics.psb.ugent.be/plaza_v1/; Proost et al., 2009) and added to the alignment. For construction of the final tree, all identified protein sequences were aligned (Table S7) using the L-INS-i option in MAFFT (Katoh et al., 2005) and edited manually. The JTT+G model was selected as the best fitting amino acid substitution model according to the Bayesian Information Criterion in ProtTest (Darriba et al., 2011). To reconstruct the phylogeny we used MrBayes 3.1 (Ronquist and Huelsenbeck, 2003) as implemented at the CIPRES portal (Miller et al. 2010) and initiated two runs of eight Markov-chain Monte Carlo (MCMC) chains of 106 generations each from a random starting tree, sampling every 100 generations (additional settings: Rates = gamma, Ngammacat = 4, Aamodelpr = JTT, Temp = 0.01). A suitable burn-in was chosen based on Tracer outputs (v. 1.5; http://tree.bio.ed.ac.uk/software/tracer/) and convergence was assessed by standard deviation 3 of split frequencies (< 0.08) and with AWTY (Nylander et al., 2008) by comparing the estimated posterior distributions of branch support from the two independent MCMC runs. The phylogenetic tree was rooted between the monocot and the eudicot sequences and visualized with the MEGA Tree Explorer (Tamura et al., 2011). Method S5. Comparison of relative UGT74 gene expression Methods for qRT-PCR experiments are described Experimental Procedures. Microarray gene expression analysis was conducted by the UC Berkeley Functional Genomics Laboratory Affymetrix Genechip Core Facility, utilizing the Affymetrix ATH1-121501 gene chip. mRNA was prepared from 2-week-old vegetative rosettes of wild-type (Col-0) plants according to their recommended protocol (http://qb3.berkeley.edu/qb3/fgl/microarray.cfm). Plants were frown asceptically according to our standard protocol for measuring glucosinolates (see Experimental Procedures). Data were downloaded for the “vegetative rosette” samples of the “Developmental Map” dataset at http://bar.utoronto.ca/efp/cgibin/efpWeb.cgi. This dataset was originally published by Schmid et al. (2005). Absolute expression values are shown. Supporting References Clough, S.J. and Bent A.F. (1998) Floral dip: a simplified method for Agrobacteriummediated transformation of Arabidopsis thaliana. Plan J., 6, 735-743. Darriba, D., Taboada, G.L., Doallo, R. and Posada, D. (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics, 27, 1164-1165. Grubb, C.D., Zipp, B.J., Ludwig-Müller, J., Masuno, M.N., Molinski, T.F. and Abel, S. (2004) Arabidopsis glucosyltransferase UGT74B1 functions in glucosinolate biosynthesis and auxin homeostasis. Plant J., 40, 893-908. Katoh, K., Kuma, K., Toh, H. and Miyata, T. (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res., 33, 511-518. Li, Y., Baldauf, S., Lim, E.K. and Bowles, D.J. (2001) Phylogenetic analysis of the UDPglycosyltransferase multigene family of Arabidopsis thaliana. J. Biol. Chem., 276, 4338-4343. 4 Miersch, O., Neumerkel, J., Dippe, M., Stenzel, I. And Wasternack, C. (2008) Hydroxylated jasmonates are commonly occurring metabolites of jasmonic acid and contribute to a partial switch-off in jasmonate signaling. New Phytol., 177, 114-127. Miller, M., Pfeiffer, W. and Schwartz, T. (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Gateway Computing Environments Workshop (GCE), pp. 1-8. Nylander, J.A., Wilgenbusch, J.C., Warren, D.L. and Swofford, D.L. (2008) AWTY (are we there yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics, 24, 581-583. Proost, S., Van Bel, M., Sterck, L., Billiau, K., Van Parys, T., Van de Peer, Y. and Vanepoele, K. (2009) PLAZA: a comparative genomics resource to study gene and genome evolution in plants. Plant Cell, 12, 3718-3731. Quiel, J.A. and Bender, J. (2003) Glucose conjugation of anthranilate by the Arabidopsis UGT74F2 glucosyltransferase is required for tryptophan mutant blue fluorescence. J. Biol. Chem., 278, 6275-6281. Ronquist, F. and Huelsenbeck, J.P. (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics, 19, 1572-1574. Schmid, M., Davison, T. S., Henz, S. R., Pape, U. J., Demar, M., Vingron, M., Scholkopf, B., Weigel, D., and Lohmann, J. U. (2005) A gene expression map of Arabidopsis thaliana development. Nat Genet 37, 501-506 Sreenivasulu, N., Radchuk, V., Alawasy, A., Borisjuk, L., Weier, D., Staroske, N., Fuchs, J., Miersch, O., Strickert, M. Usadel, B., Wobus, U., Grimm, B., Weber, H. And Weschke, W. De-regulation of absisic acid contents causes abnormal endosperm development in the barley mutant seg8. Plant J., 64, 589-603. Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M. and Kumar, S. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol., 28, 27312739. 5