Genetics chapter 7 questions
advertisement

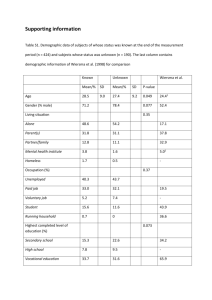
Genetics chapter 7 practice questions 1) A 3 year old male presents with behavioral problems, dysostosis multiplex, and mental retardardation. After running several tests it is determined that he has a deficiency of NAcetylglucosamine-6-sulfatase. Which of the following mucopolysaccharidoses does he have? a) Hunter b) Morquio A c) Morquio B d) Sanfilippio C e) Sanfilippio D 2) A patient has been diagnoses with a lysosomal storage disorder. They have had their DNA analyzed and they have one N370S allele likely impacted their prognosis. Which of the following disorders do they likely have? a) Gaucher type 1 b) Krabbe c) Sandhoff d) Schindler e) Tay-Sachs 3) A patient with a urea cycle disorder suffers from a progressive condition of spastic quadriplegia and mental retardation. Which enzyme deficiency does the patient suffer from? a) Arginase b) Argininosuccinase c) Argininosuccinic acid synthase d) Carbamyl phosphate synthase e) Ornithine transcarbamylase 4) While on rotations you see a 7 year old boy who is awaiting a kidney transplant. You see that he has a history of corneal crystals, electrolyte disturbances and severe renal glomerular damage due to crystals accumulating in the lysosomes. What metabolic disorder does this boy likely have? a) Cystinosis b) Cystinuria c) Hereditary Fructose Intolerance d) Hereditary Tyrosinemia type 1 (HT1) 5) A histologic slide is presented at a grand rounds. In the cells are an accumulation of oligosaccharides, lipids, and glycosaminoglycans. The tissue sample came from a 6 year old child with coarse facial features, skeletal abnormalities, hepatomegaly, corneal opacity and mental retardation. The child has a mutated gene encoding mannose-6-phosphate recognition molecules. What disease will likely cause this child’s early death? a) I-Cell disease b) Maple syrup urine disease c)Pompe disease d) Zellweger syndrome 6) X-linked Adrenoleukodystrophy is a peroxisomal disorder involving faulty Beta-oxidation of very long chain fatty acids (VLCFA). A patient with the condition comes to your office. Looking at your chart you see that the patient has been categorized as falling under the adrenomyeloneuropathy (AMN) subdivision of the disorder. Which of the following patients are you most likely treating? a) a 3 year old heterozygous male b) a 3 year old homozygous male c) a 3 year old heterozygous female d) a 27 year old heterozygous female e) a 27 year old homozygous male 7) A mother at 27 weeks gestation goes into preterm labor following a health crisis involving her liver. The infant is tested for a lipid metabolism disorder following delivery. Which disorder in fatty acid oxidation was the fetus most likely affected with. a) AFLP b) LCAD c) LCHAD d) MCAD e) SLO Answers: 1) Answer is E. Page 140-1 of Medical genetics. Out of all of the Mucopolysaccharidoses conditions only Hurler, Hunter and Sanfilippo are characterized by mental retardation, the rest have normal cognition. Therefore even though Morquio A and Sanfilippio D both have mutant NAcetylglucosamine-6-sulfatase, Morquio A does not match the symptoms. Here is a list of associated mutant enzymes with each condtion: a) Hunter - Iduronate sulfatase b) Morquio A - N-Acetylglucosamine-6-sulfatase c) Morquio B - Beta-Galactosidase d) Sanfilippio C - Acetyl-CoA:alpha-glucosaminide N-acetyltransferase e) Sanfilippio D - N-Acetylglucosamine-6-sulfatase 2) The Answer is A. Page 142 Medical Genetics. Gaucher disease has traditionally been divided into three subtypes on a wide spectrum of clinical presentations. Type 1 is most common and least severe, without involvement in the CNS. Type 2 is most severe and often leads to death before 2. Type 3 is somewhere between those other two types. “Persons with a least one N370S allele... do not develop primary neurological disease and tend to have a milder outcome in general.” 3) Answer is A. Page 143-4 Medical Genetics. Clinical presentations of B-E deficiencies all have very similar clinical presentations. This is because they all lead to a buildup of urea precursors. Symptoms are progressive lethargy and coma. Arginase however causes the patient’s symptoms, it would appear based on the chart on page 144 that this deficiency would lead to a buildup of arginine. 4) Answer B. Page 132, 136, 145 Medical Genetics. Cystinosis is a rare disorder caused by reduced ability to transport cystine across the lysosomal membrane, This results in crystal accumulation in the lysosomes of most tissues. Renal glomerular damage is severe. Cystinuria is a dibasic amino acid transport disorder that results in increased cystine, lysine, arginine and ornithine levels in the urine. This too can result in eventual renal failure, but is more commonly associated with renal calculi due to the insolubility of cystine. Hereditary Fructose Intolerance often presents with poor feeding, failure to thrive, renal and hepatic insufficiency, there are no crystals implicated in this disease. Hereditary Tyrosinemia type 1 (HT1) is characterized by dysfunction of the renal tubules, acute episodes of peripheral neuropathy, progressive liver disease leading to cirrhosis and a high risk for developing liver cancer. It is associated with accumulation of fumarylacetoacetate and maleylacetoacetate, but not with accumulation of crystals 5) Answer A. Pages 134, 136, 140, 142-3 Medical Genetics. I-Cell disease results from a deficiency in mannose-6-phosphate recognition markers that are posttranslationally attached to enzymes to mark them for targeting to transport them into the lysosome. Therefore these is an accumulation in the cytoplasm of fibroblast cells of oligosaccharides, lipids, and glycosaminoglycans, because they cannot enter the lysosome. Maple syrup urine disease does result in early death, is associated with an accumulation of branched-chain amino acids. Zellweger syndrome also results typically in infant death.This is a peroxisomal disorder which is involved in degradative metabolic functions related mainly to lipid metabolism. However it is characterized by severe hypotonia in neonates, progressive disease of the white matter or the brain, and a distinctive facial appearance. Pompe disease is a glycogen storage disease that mainly affects muscle tissue including the heart, causing myopathy and sometimes early death. 6) Answer D. Page 140 Medical Genetics. The AMN subdivision of the disease is similar to the Childhood Cerebral ALD (CCALD) subdivision. CCALD onsets typically around 3-10 years of age and leads to progressive cognitive and behavioral deterioration. AMN causes similar less severe neurological symptoms. It has a much later age of onset and a slower rate of progression. A-C is wrong because the 3 year olds are too young to show symptoms if they were diagnosed with AMN. E and also B is wrong because as indicated by the name of the disease it is an X-linked condition, males can never be homozygous they only have one X chromosome. Most diseases of X-linked inheritance produce females who are asymptomatic carriers. However, 40-50% of female carriers of X-linked Adrenoleukodystrophy have AMN symptoms. 7) The Answer is C. Page 137-8 Medical Genetics. LCHAD is Long Chain L-3-hydroxyacyl-CoA dehydrogenase is the most severe Fatty acid oxidation defect. It is an enzyme in the complex of Mitochondrial trifunctional protein. In a woman pregnant with an affected fetus it can cause Acute fatty live of pregnancy, AFLP as well as Hemolysis, Elevated Liver function tests, Low Platelets, HELLP syndrome. Preterm deliveries are common.