File - South Waksman Club
advertisement

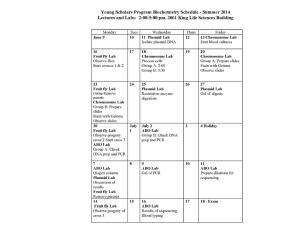
Waksman ’11-‘12 MacCallum Robertson DSAP Walkthrough DSAP stands for DNA Sequencing Analysis Program. What this program does is basically does is allow us to analyze a specific DNA sequence from Wolffia australiana. When we complete analyzing the sequence, we send in our results to Rutgers, who then send it to NCBI to be published. ~The way this works (background info) is that the DNA that is about to be sequenced, the template DNA, is prepared as singled strand DNA. A primer (an oligonucleotide) is attached to both ends of the template DNA which will allow for the synthesis of a complimentary strand of DNA. Four reactions using the same template DNA, will be needed (one for each base). What occurs is that the once a deoxynucleotide is added to the template, DNA polymerase moves along the template strand and continues to add bases until a dideoxynucleotide is added (only a small amount of these are added to the mixture and allow for the reaction to end at random times and with billions of strands of DNA produced, there will be strands of different lengths, all ending with a dideoxynucleotide. When these are transferred to a gel, the shortest strands reach the bottom first and the dideoxynucleotide, which are tagged with a fluorescent dye, give off light and is read by a sequencer. Therefore, the DNA will be “reconstructed” from looking at the color of the dideoxynucleotide and the corresponding nucleotide. So for example, if the smallest strand ended with the dideoxynucleotide Adenine, and the second smallest ended with Thymine, they would move to the bottom of the gel the fastest and be sequenced (based on their tags) in the order Adenine, Thymine. This is done with billions of strands to ensure all the base pairs are recorded. ~Recently, a more efficient method called “Shotgun sequencing” has been adopted by Rutgers, in which longer strands of DNA are broken up into fragments and reassembled after the procedure above is completed. (All of this is automated). Now back to DSAP. Well after all that happens, we get a waveform, which is the sequence you download from DSAP for each clone. The waveform is the sequence and the actual waves represent the wavelength of the fluorescent tags mentioned above, which tell us which nucleotide it is. Sometimes, this will not be readable, meaning you are looking for DEFINIED peaks, not a jumbled up mess. If it is really messy, it can be classified as “unreadable” ~This would be considered a readable waveform. These waves represent the wavelengths of the fluorescent tags and therefore the base pairs (the letters above the waves) Waksman ’11-‘12 MacCallum Robertson How to start: The first thing to do is find (easiest to use the find bar in Finch TV) the part of the sequence that is GGCCGGG. Once you find this, look for the first nucleotide that is not a G (so there may be more than 3 G’s) and that is the start of your sequence, for example, if you have GGCCGGGGGGA, A will be where your sequence starts. You will have to cut everything to the left of this first base pair BUT when DSAP asks what nucleotide your sequence starts at, you will have to put the base and the original number in the sequence. This number can be found above the nucleotide letter, once you cut it, the first base becomes 1, you will need the number it was before cutting, for example A51. Now to find the end of the sequence, look for the Poly A tail, a chain of Adenine nucleotides (which ultimately protect the mRNA when it leaves the nucleus of a cell). Once you find this poly A tail (in addition to a chain of A’s if you look at the waveform it will look like a bunch of green waves higher up compared to others) cut the everything AFTER the first non-A base, for example: AAAAAAAAATA, you would cut everything starting at, and including, T. NOTE: You may see that in there may be a base in what seems to be the middle of a Poly A tail, meaning that there is the same raised green waveform on both sides, then it may have been a sequencing error, and the base can be changed to and a. Also, if the sequence contains a decent amount of readable base pairs, around 400-500, and it becomes unreadable, just crop it off where it becomes unreadable. Lastly, if the Poly A tail is missing, just crop it at the end of the sequence or where it stops being readable. Edit Sequence: In this section, you will have to go back to your sequence and add in any missing nucleotides or ambiguous nucleotides (represented by “N”). You will be able to figure out what these nucleotides are by looking at the waveform, whichever wave is the largest is the correct nucleotide. Blasts: What you are really doing when you “Blast” a sequence is comparing your sequence with all of those found on a cDNA database. “Blast N” is a comparison based on the nucleotide sequences (hence the “N”) in which you get back matches organized by how closely the sequences match. When they ask you to switch to the “est database” it is just to cross reference with another database. ~ You can tell how close your sequence is to another by looking at the E value. The smaller the E value, the closer that sequence is to your sequence, and a “good” E value is considered anything below e^-9 (there is some leeway with 0.0 being the best, meaning both sequences are the same. ~The accession #, definition, and organism can all be found by clicking on the different sequence matches (found under the bar graph). This bar graph essentially shows you the relative abundance of sequences that are closely related to yours, so more red line (red lines meaning that a 200+ nucleotides match). One important part of the Blast is the Query. DSAP will ask you query start and end, and to find this all you have to do is scroll down past all the sequence matches until you see the actual nucleotide sequences (which match the order of the sequences represented by their organism and accession number above). You will just have to look at the part that says “query” (the top set of nucleotides, above “sbjct”) and see where it starts and ends, for example: Start at A53 and end at T803. What this means is that the sequence that matches yours starting at wherever the query begins and ends wherever the query ends. Blast X: This is the same idea as a Blast N but this references an amino acid database. For Blast X, the query shows the matching proteins (this is found by translating the nucleotides into Waksman ’11-‘12 MacCallum Robertson proteins by looking at codons, or triplets of nucleotides, and the proteins that correspond to them). One important thing mentioned right above the query is the “Frame” which will say either Frame=+1, +2, or +3. What this tells you is the most probably reading frame. ORF (Open Reading Frame): In this step you will try to find the sequence of a working protein that corresponds to your sequence (so you can use it in Blast P, matching your sequence to other protein sequences). When you copy your sequence into the “Toolbox” there will be the option of 3 different reading frames. These reading frames are different ways of looking at your sequence. Three nucleotides are considered a codon, which code for amino acids, so when you look at a sequence like AAGGCC, you could find the first amino acid correspond to AAG. The point of a reading frame is that this may not be the correct amino acid, so it finds the amino acid corresponding to the sequence as if you started at the second nucleotide (hence reading frame +2). This would be AGG, and could code for a different amino acid. Then lastly, there is reading frame +3, which starts on the third nucleotide. The reason that there are only 3 reading frames is that the pattern would repeat (codon will be the same). NOTE: The most probably reading frame can be found in the Blast X, above the query (mentioned above). Analyzing ORF’s: Once you convert your sequence into proteins (blue and underlined) and have found the correct reading frame, you must look for the start of the protein, which is characterized by the letter M. M stands for Methionine (the corresponding codon is AUG) which always starts a protein sequence, just as it always ends with one of a few stop codons (UAG, UGA, and UAA) which is found for you and represented by “*”. To get your amino acid sequence, copy the letters starting from Methionine to the *, (the * after the chain of blue underlined proteins), not including the *. ~Notice how each frame has its first base (#1) correspond to a different spot. This represents the different reading frames, all starting from shifted points on the sequence. Waksman ’11-‘12 MacCallum Robertson UTR (Untranslated Region): The untranslated regions can be found via the toolbox. These regions are simply parts of the sequence that whose amino acids do not correspond to a of proteins, hence untranslated. One can find this by looking at the toolbox, and whatever amino acids are not highlighted in blue are considered untranslated. All sequences before the AUG (Methionine) is the 5' UTR and all the sequences after the stop codon is a 3' UTR. There is a distinction between the untranslated region before and after the translated region, and that is the name, either 5’ UTR or 3’ UTR. The 5’ UTR is the region before the translated one and the 3’ is the region after. This is because translation (coding for amino acids and therefore proteins) is in the 5’ to 3’ direction of DNA. Analysis: 1.) The first question DSAP asks you is what the function of your protein is. From the results of your blasts you should be able to be able to have a general idea of the proteins function, and if you don’t know what it does, Google, Wikipedia etc, the term or protein. 2.) The second question just asks you if there is a similar protein in humans, which you find by going to one of your Blasts and look in the results for Homo sapiens (Use a find feature like “Ctrl F”). As for what this tells you about the protein, you can answer with a multitude of responses like “the function of the protein is something utilized by both mammals and plants alike” or something of the sort. 3.) Question 3 asks you if your protein contains any domains. A protein domain is a structure (part of the protein) that can exist and has a specific function. Each domain has its own function and oftentimes proteins are composed of multiple domains, such as Pyruvate Kinase (picture to the left), which has 3 domains, represented by the three different colors. To find these domains you can go to your Blast P and click on the large red bar at the top (the protein super family: a group of proteins with similar functions) and there will be a list of protein domains. Hover your mouse over one of these and it will tell you the name and function. 4.) For question #4, DSAP asks if there exists a 3Dimensional structure of a protein similar to yours. The way you find this structure is to go the same place as in question 3, the list of protein domains, and click on one of the choices. There will be a PubMed page with a 3D image of the protein along with the accession number (identification #) on the left of the page. Waksman ’11-‘12 MacCallum Robertson 5.) Question 5 is slightly longer than most of the others but all you have to do is list the answers out. This question only applies if there was a 3Dimensional structure of a homolog (protein with a similar function) to your protein. Part “a” asks you for the accession number of the protein, which you found in question 4. Part “b” asks you how many alpha helices there are in the protein. An alpha helix is a reoccurring structure in the secondary structure (general 3D structure) which is represented by cylinders in the 3D rendition. Part “c” it asks about the presence of beta-strands, which form beta-sheets (polypeptide chains with intermittent Hydrogen bonds) with other Beta-strands, within the homolog. Beta-strands, like alpha helices, are a reoccurring structure found in the secondary structure of a protein that a represented by rectangular prisms. In part “d”, when they refer to parallel and antiparallel they are referring to the orientation of the peptide strands that comprise the beta-sheets. If it is parallel, the peptide strands are running in the same direction while in the antiparallel betasheet they run in opposite directions. The way to tell this on the 3D model is by looking at the orientation of the betastrands, if they look like they are angled in such a way that their slope is positive, then they are parallel, if they have a negative slope, they are antiparallel. ~ A 3D structure of a protein (Patatin) ~The purple structures are alpha helices and the orange structures are antiparallel beta strands Waksman ’11-‘12 MacCallum Robertson 6.) A cofactor is a non-protein chemical compound (organic or inorganic) that binds to a protein and is necessary for the protein to perform its function. Oftentimes the protein that have cofactors are enzymes, hence cofactors commonly being classified as coenzymes. A substrate is similar to a cofactor in the fact that it pertains to an enzyme but a substrate is a molecule that an enzyme is acting on. Lastly, proteins also bind to other proteins in order to carry out their biological function. You will be able to determine if a 3D structure of the protein is binding to one of the previously mentioned items simply by looking to see if something is attached to the protein itself. Oftentimes the 3D structures are not bound to another structure. ~A protein and a cofactor 7.) The last question depends the function of your protein and how creative you feel like being. Your answer must consist of a potential experiment that could successfully determine the function of your protein, for example if your protein has anti-fungal functions, then you could design a test in which two organisms are exposed to fungi, one with the protein and one without, and observe the effects on both organisms. Another potential answer is suggesting “gene silencing” in which a gene is “switched off” and the change in the organism, when the gene is not being expressed, can be seen and from that the function can be determined. SUBMIT ( ALMOST THERE :D ) ~ All you have to do is “characterize” the clone, it will either be coding or noncoding (when it is unreadable). Finally you will have to sum up the protein’s function in one sentence. Once you have finished Practice Clone #’s 1-4 you can get your own clones (do this by going to “My Clones” and in the top right there will be something that says “+ Add New Clone” to sequence and GET PUBLISHED!