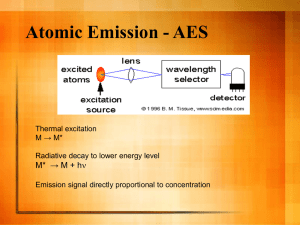
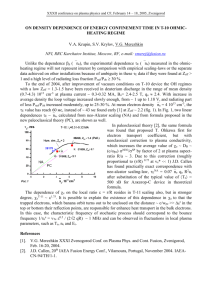
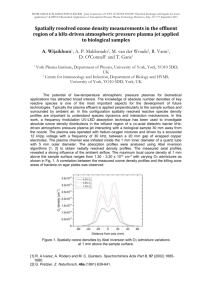
LC-MS/MS Analysis
Instrumentation
Quantification of PAESe was performed using an Agilent Technologies, Inc., XCT Ultra
Plus ion-trap mass spectrometer with an ESI interface (Santa Clara, CA), following
chromatographic separation using an Agilent 1100 HPLC system consisting of an autosampler
and binary pump. Solvents were vacuum filtered through a 0.45 m nylon membrane (Gelman
Sciences, Ann Arbor, MI, USA), and degassed prior to use. Separation was achieved by
automated injection of 20 L samples onto a reversed phase C18 guard and analytical column
Zorbax Eclipse Plus C18 column (100 × 4.6 mm, 3.5 μm, Agilent Technologies) equipped with a
Phenomenex (Torrance, CA) Security Guard C18 guard column (4.0 × 3.0 mm) under isocratic
conditions at a flow rate of 250 L/min. An additional in-line pre-filter (MacMod Analytical,
Inc., Chadds Ford, PA, USA) was inserted before the guard column. The column effluent was
introduced into the mass spectrometer using an electrospray ion-source. High purity nitrogen was
used as the curtain and collision gas. Data analysis was performed using ChemStation, v. 6.1
(Agilent Technologies, Santa Clara, CA).
MS Optimization
PAESe and FPAESe, 4-fluoro-phenyl-2-aminoethyl selenide - used as internal standard
(I.S.), were dissolved at a concentration of 0.5 g/mL in mobile phase consisting of MeOH and
0.2% (v/v) formic acid (50:50, v/v) and filtered through a 0.45 m nylon filter (Gelman
Sciences, Ann Arbor, MI). Samples were introduced into the mass spectrometer via the
ionization source at a flow rate of 6.0-10.0 L/min using a syringe pump (kdScientific Inc., New
Hope, PA). Parent compounds were identified from total-ion chromatograms (TIC) acquire in
positive ion-mode over the range of 100-1000 m/z. Product-ion chromatograms (XIC) were
acquired for each parent (precursor) ion (i.e., PAESe and FPAESe) over the range of 50-200 m/z.
A unique ion-pair (precursor and product ion) was identified for each compound. Mass
spectrometry parameters for individual ion-pairs were optimized initially using manufactures
software and then refined in the manual tune mode. Desolvation gas was set at 350 °C with a
flow rate of 9.0 L/min and nebulizer pressure of 40.0 psi. The optimal capillary voltage and
capillary exit voltage were 4000 V and 100 V, respectively, with a skimmer voltage of 40 V.
Preparation of Standards and Quality Control Samples
Stock solutions of PAESe and FPAESe were weighted individually and dissolved in
mobile phase to prepare stock solutions for use as standards, quality control (QC) or I.S. at 100
μM. To ensure similar sample matrices, calibration curves were prepared by serial dilution of a
PAESe stock solution into mobile phase. A 10 L volume of each was transferred into a vial
containing 10 L FPAESe (100 M), 50 L blank plasma and 180 L ACN to achieve a plasma
concentration range of 2,000, 1,600, 1,000, 400, 200, 160, 100, 50 and 20 nM. Quality control
(QC) samples were prepared in triplicate at 60, 300 and 600 nM, as described above. The
FPAESe stock solution was diluted in mobile phase to achieve a final concentration of 400 nM.
All standard and QC samples were prepared freshly for each analysis.
Sample Preparation
Plasma samples (50 μL) were deproteinated with ice-cold ACN (200 μL), to achieve a
final plasma/organic solvent ratio of 20% (v/v), and vortexed to precipitate plasma proteins. A 10
μL volume of freshly prepared FPAESe solution was diluted in mobile phase then spiked into the
mixture of ACN and plasma to achieve a final concentration of 400 nM. The mixture was cooled
in an ice-water bath for 1 hr, centrifuged at 1,200 × g for 10 min at 4°C. The deproteinized
supernatant was dried with a stream of filtered nitrogen and the residues were reconstituted with
100 μL of mobile phase by vortex mixing, resulting in a 50% dilution of samples and standards.
Reconstituted samples were transferred into low volume glass vial inserts for LC-MS/MS
analysis.
Assay Performance and Validation
Standard and quality control samples were prepared by extraction of pooled plasma from
untreated animals, described above. The specificity and selectivity of method was determined
based on MRM response of drug-free plasma and plasma spiked with PAESe or FPAESe to
monitor specific ion-transitions and the retention time of the analytes of interest. Standard curves
for PAESe were constructed by calculating the ratio of the analyze peak area to that of the I.S.,
and plotting the ratio versus the theoretical concentrations. Data was fit using weighted least
squares; the inverse of the variance (1/x2) of the data was used as a weighting factor. The
standard curve was considered acceptable if the calculated accuracies of >90% of the standards
were within 15% of their theoretical value and no systematic deviations over the linear range
were observed. The LLOQ was determined for the lowest standard with an accuracy within
±20% of the theoretical concentration with a coefficient of variation (CV) < 20% and a signal
intensity that was at least 5 times greater than the baseline noise (S/N > 5). The absolute
sensitivity was determined experimentally by calculating the amount of analyte that was applied
to the column at the LLOQ, given the sample injection volume (20 L). Intra-day and inter-day
variation and accuracies were determined by analyzing QC samples, in triplicate, prepared at 3
different concentrations independently within 24 hr or at 3 different concentrations each day over
3 or more separate days within a 7 day period.
Sample stability was determined under various temperature-dependent storage
conditions. Two different concentrations of PAESe (0.5 and 1.0 μM) in triplicate were stored at
room temperature, -20 or -80°C for 4 weeks. For freeze-thaw stability, the samples were stored at
-20°C, and were then thawed at room temperature for 24 hr followed by refreezing at -20°C for 3
cycles. Stability was determined by comparing the measured concentration of each sample to
theoretical concentration of freshly prepared samples; a percent change or CV < 15 % was
considered significant.
Validation of LC–MS/MS Method
The representative total ion-chromatogram (TIC) and product ion-chromatogram (XIC)
of plasma sample spiked with PAESe or FPAESe are shown in Fig. S2 and S3. A unique
precursor/product ion-pairs at 184.0 → 154.7 m/z for PAESe and 204.0 → 174.7 m/z for I.S.
were identified. A retention time of 3.6 min for both PAESe and FPAESe with a total
chromatographic time of 5.0 min was achieved. Neither cross-talk, nor endogenous interference
around the retention times of PAESe and FPAESe in the blank plasma were observed.
The LLOQ of PAESe was 50 nM with inter-day accuracy of 80.8% and a precision of
18.2%, which was sufficient for the purpose of this study. A good linearity was shown over the
entire concentration range with a correlation coefficient (r2) of 0.995 or better. Intra- and interday accuracies and precision of the QC samples were within an acceptable range of ±15% of
their theoretical concentrations (Table SI). No significant degradation at 20 (room temperature),
-20 or -80 oC was observed; all stability samples ranged from 98 to 110% of their theoretical
concentrations with CV of 3.43 to 13.0%.
Table SI. Intra-day and Inter-day accuracy and precision for PAESe
Intra-day assay
Inter-day assay
QC
Accuracy
Precision
Accuracy
Precision
(nM)
(%)
(CV%)
(%)
(CV%)
60
101
14.0
102
14.9
300
103
8.5
95.9
14.4
600
93.3
10.4
95.9
9.8
Legends for Supplementary Figures
Fig. S1. Encapsulation Efficiency according to the drug:lipid ratio Encapsulation efficiency
of SSL-PAESe was determined by altering drug:lipid ratio. Encapsulation efficiency was
decreased with increasing drug:lipid ratio.
Fig. S2. Total ion chromatogram (a) PAESe; (b) FPAESe (I.S.) Blank rat plasma spiked with
PAESe or FPAESe of 300 ng/mL was deproteinated with ACN to remove plasma protein. The
mixture was cooled on ice for 1 hr, and then was centrifuged at 1,200 × g for 10 min at 4°C. The
supernatant transferred into new tubes was evaporated with a stream of dry nitrogen, followed by
reconstitution with mobile phase to inject into the LC-MS/MS system for analysis. The retention
time was 3.6 min for both PAESe and FPAESe.
Fig. S3. Product ion chromatogram (a) PAESe; (b) FPAESe (I.S.) Blank rat plasma spiked
with PAESe or FPAESe of 300 ng/mL was prepared as described in Fig. S2, MRM mode was
applied to monitor specific ion transitions of PAESe and I.S. The maximum signal was detected
as the precursor ions at m/z 184.0 and m/z 204.0 for [M+H]+ of PAESe and I.S., respectively.
The most intense signal was determined at m/z 184.0 → 154.7 for PAESe and m/z 204.0 →
174.7 for I.S. as precursor/product ion-pairs.
100
80
EE %
60
40
20
0
0.2 : 1
0.5 : 1
1:1
Drug:Lipid
Fig. S1.
2:1
(a)
(b)
Fig. S2.
Intens. 3.
x10 4
+MS3(184.0->i155.0), 3.2-3.8min #(87-103)
(a)
1.0
154.7
0.8
0.6
0.4
0.2
0.0
50
Intens. 2.
x10 4
75
100
125
150
175
200
225
250
275
m/z
+MS3(204.0->i174.0), 3.4-3.8min #(92-104)
(b)
2.5
174.7
2.0
1.5
1.0
0.5
0.0
50
75
100
125
150
175
Fig. S3.
200
225
250
275
m/z
 0
0