Module 7 - Protein Structure Prediction
advertisement

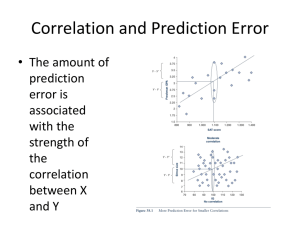
MODULE 7 Protein Structure Prediction AIMS To understand how computer algorithms can be used to predict the secondary and tertiary structures of proteins To recognize different approaches to structure prediction To understand some aspects of the limitations of computer-based methods OBJECTIVES The student should be able to: Predict the occurrence of aspects of secondary and tertiary structure in proteins using Web-based analytical tools To select which tools are most appropriate for a particular analysis INTRODUCTION Protein structure may be considered at a variety of levels (for further information see Webbased tutorial): 1o (primary) structure is the actual amino acid sequence of the protein 2o (secondary) structure refers to the localized organization of parts of the polypeptide chain (e.g. helix, sheet, turn etc.) 3o (tertiary) structure describes the three-dimensional organization of all the atoms in the polypeptide 4o (quarternary) structure refers to the organization of a protein composed of more than one polypeptide chain This module deals with the prediction of the secondary and tertiary structure of proteins. The most direct route to the study of protein structure is the use of techniques such as X-ray crytallography and NMR to determine the atomic co-ordinates of a protein. However, whilst there are over 100,000 entries in the primary protein sequence databases, there are only just over 12,000 entries in the protein structure databases. In consequence, a variety of methods are in development to predict secondary and tertiary structure from the 1o sequence information and this is the topic covered by this module. In truth this is an enormous subject worthy of a course all to itself, so only a somewhat superficial view can be presented here. More detailed tutorials and guides, such as “Sisyphus and protein structure prediction”, “Pedestrian guide to analysing sequence databases”and “A Guide to protein structure Prediction”, are available on the Web. Secondary structure prediction The most successful area of protein structure prediction deals with secondary structure and related topics including the interaction of proteins with membranes. Signal peptides Signal peptides (or signal sequences) are short N-terminal amino acid sequences that target the protein for membrane translocation and are removed after translocation. SignalP predicts signal peptide cleavage sites in Gram-positive, Gram-negative and eukaryotic amino acid sequences. http://www.cbs.dtu.dk/services/SignalP/caution.html Intracellular targeting TargetP predicts the subcellular location of eukaryotic protein sequences. The subcellular location assignment is based on the predicted presence of any of the N-terminal presequences chloroplast transit peptide, mitochondrial targeting peptide, or secretory pathway signal peptide Trans-membrane helices Many proteins in the cell are integral membrane proteins that have one or more segments embedded in. In transmembrane proteins one or more segments of the protein completely traverse the phospholipid bilayer and these membrane spanning domains are always helices or multiple strands. Arguably, the most successful area in secondary structure prediction is that of the prediction of trans-membrane helices. There are a variety of computational approaches which offer 90% accuracy or more in such predictions. We will focus on one of the approaches, known as TMHMM, although there are others such as TopPred2, MEMSAT, DASand PHDhtm, which you might have a look at. The large majority of trans-membrane helices consist of an unusually long stretch of hydrophobic amino acid residues and it is this feature that many programs employ to identify such potential helices. The helix also has a topology i.e. whether it runs inwards or outwards. Positively charged residues, arginine and lysine, play a central role in determining the orientation since they are primarily found in non-transmembrane parts of the polypeptide on the cytoplasmic side. TMHMM employs a hidden Markov model which closely onto these features to make highly accurate predictions of trans-membrane helices. Have a look at the output from a typical TMHMM analysis of the lactose permease (LacY) from E. coli. Notice it has 12 predicted trans-membrane helices with their polarity clearly indicated. helices and sheets etc. One of the first predictive algorithms GOR (Garnier, Osguthorpe & Robson, 1978) for secondary structure was developed through a co-operation between a laboratory interested in developing the theory for protein secondary structure prediction methods and a laboratory interested in applying and comparing such methods . The GOR algorithm unambiguously assigns each residue to one conformational state of a-helix, extended chain, reverse turn or coil. In its initial form GOR was roughly 50% accurate on a test sample of 26 proteins. GOR has now been through a series of developments and version IV of GOR has a mean accuracy of 64.4% for a three state prediction. The program gives two outputs, one eye-friendly (example) giving the sequence and the predicted secondary structure in rows, H=helix, E=extended or beta strand and C=coil; the second (example) gives the probability values for each secondary structure at each amino acid position. The predicted secondary structure is the one of highest probability compatible with a predicted helix segment of at least four residues and a predicted extended segment of at least two residues. There are a number of other secondary structure prediction approaches including PSIPRED, PHD, NNSP, PROF, Predator and ZPRED. Most of these servers expect the input to an alignment of multiple sequences which enhances the accuracy of the predictions. Jpred developed as a result of a study to test and compare different secondary structure prediction methods. Jpred takes a single input sequence and scans it against a non-redundant sequence database. The hits are aligned with CLUSTALW (v1.7) and the alignment is submitted to MULPRED, which uses a combination of single sequence methods that are combined to give a prediction profile, from which a consensus is taken. The methods used within MULPRED are Lim, GOR, Chou-Fasman, Rose and Wilmot/Thornton turn prediction methods. The accuracy of Jpred is approximately 73%. Super-secondary structure Secondary structure elements are observed to combine in specific geometric arrangements known as motifs or super-secondary structures (see Web-based tutorial) e.g. coiled coils, helix-turn-helix etc. Coiled-coils are another structural feature of proteins which sometimes separate domains. Coiled coils comprise two, three or four amphipathic helices wrapped round one another. Coiled coil motifs are particularly amenable to computer-based prediction because of the characteristic repeating patter of hydrophobic residues spaced every four and then three residues apart. This pattern forms a heptad repeat (abcdefg)n of amino acids in which positions a abd d tend to be hydrophobic and positions e and g are predominantly charged residues. Predictions of coiled coils can be obtained at PAIRCOIL and MULTICOIL. The leucine zipper structure is adopted by one family of the coiled coil proteins. Leucine zippers have a characteristic leucine repeat: Leu-X6-Leu-X6-Leu-X6-Leu (where X may be any residue) and TRESPASSER will detect such motifs with a high degree of accuracy. The helix-turn-helix motif occurs in many DNA binding proteins and can be predicted using HTH. Integrated structure prediction There is a variety of servers which offer a secondary structure prediction integrated with a variety of other analyses. Predict Protein offers predictions of: secondary structure (more info), solvent accessibility (more info), globular regions ( more info), transmembrane helices (more info), coiled-coil regions ( more info). as well as a multiple sequence alignment (i.e. database search), ProSite sequence motifs (more info), low-complexity retions (SEG) ( more info), ProDom domain assignments (more info), Tertiary structure prediction This component of the module, more than any other, can only skim the surface of a complex and extensive topic. An excellent and more detailed introduction to the the topic is provided in “A Guide to Structure Prediction (version 2)”. The ultimate objective in protein structure prediction is to use ab initio methods to accurately predict the tertiary structure of a protein from its primary structure using purely physicochemical information. However, such approaches are prevented at present by a lack of some of the basic information required combined with the enormous computational complexity of the task. Tertiary structure describes the folding of the polypeptide chain to assemble the different secondary structure elements into a particular arrangement. Just as helices, sheets etc. are the units of secondary structure so the folds/domains are the units of tertiary structure. In multidomain proteins, tertiary structure includes the arrangement of domains relative to each other as well as the arrangement of residues within the domain. The terms ‘domain’ and ‘fold’ to a large extent mean the same thing though definitions may vary. Domains are regions of contiguous polypeptide chain that have been described as compact, local, and semiindependent units. A fold is defined as a component of tertiary structure in which the proteins have the same major secondary structures in the same arrangement with the same topological connections. There are glossaries of the different protein folds/domains. The overall strategy for secondary structure prediction is summarized by the following flowchart An excellent, more detailed, interactive flowchart has been produced by Robert Russell. The first step in any attempt to predict the tertiary structure of a protein is to search the sequence databases for proteins that show sequence similarity. If the result of the search includes a protein of known structure then the route of choice is homology modelling. If there is no homologue in the structural databases then things become rather more difficult, but not impossible. Even with no no homologues of known structure it may be possible to use fold recognition methods. There is a so called “twilight area” of 20-30% sequence identity, where it is difficult to assess whether One of the most important advances in sequence comparison recently has been the development of both gapped BLAST and PSI-BLAST (position specific interated BLAST). Both of these have made BLAST much more sensitive, and the latter is able to detect very remote homologues by taking the results of one search, constructing a profile and then using this to search the database again to find other homologues Homology modelling The most successful tool for prediction of 3D structure is homology modelling. An approximate 3D model can be built for a protein, if it has “significant similarity” to a protein of known structure. So what is “significant similarity”? The answer is about 30% identity. At this level of identity it is possible to construct a model which has a correct fold structure, but may have inaccurate loops. Above levels of 90% sequence identity, homology modelling is about as accurate as the experimental determination of a protein structure. Part of the problem of homology modelling at lower levels of similarity is to correctly align . Sequence alignments are more or less straightforward for levels of above 30% pairwise sequence identity. The region between 20 and 30% sequence identity is frequently referred to as the twilight zone. Fold recognition It has long been recognised that proteins often adopt similar folds despite lack of significant sequence or functional similarity. Fortunately, certain folds crop up time and time again in proteins, and so fold recognition methods for predicting protein structure can be very effective. Methods of fold recognition attempt to detect similarities between the 3D structure of proteins that do not exhibit significant sequence similarity. There are numerous different approaches to fold recognition, though ‘threading’ is a common feature of several of them. Some fold recognition programs can be accessed through the Web e.g.TOPITS, and 3DPSSM. If you have predicted that protein under study contains a particular fold then it is important to establish which other proteins that contain a similar fold by looking at databases such as SCOP (Structural Classification of Proteins) or CATH (Protein Structure Classification). Threading Threading takes the query sequence of unknown structure threads it through the atomic coordinates of a protein whose structure is known. The query sequence is moved residue by residue through the template sequence and calculations are carried to determine the degree of “fitness” of the alignment by a variety of methods which could include thermodynamic criteria, solvent accessibility, secondary structure information etc. Such approaches are quite computationally intensive, but there are freely accessible Web-based sites which will carry out a threading analysis e.g. bioinbgu. Building the model Sophisticated and usually expensive software is commercially available for carrying out tertiary structure predictions, but there is a freely accessible Web-based modelling server. SWISS-MODEL is an Automated Protein Modelling Server running at the GlaxoWellcome Experimental Research in Geneva, Switzerland. When a sequence is submitted to SWISSMODEL the sequence of events is as follows: 1. BLASTP2 finds all similarities of target sequence with sequences of known structure. 2. Templates with sequence identities above 25% and projected model size larger than 20 residues are selected. This step also detects domains which can be modelled based on unrelated templates 3 ProModII then generates the models in which the key process is the production of a framework which represents topology of corresponding atoms in the query sequence and the template(s). 4 Energy minimisation analysis is done for all models CPHmodels is another Web based homology modelling server. Exercises 1. Use TMHMM to predict whether the human integrin beta subunit is likely to be an integral membrane protein and, if so, how many trans-membrane domains it has. 2. What advantages might TMHMM have over TopPred (see the original TMHMM paper) 3. Use GORIV to do a secondary structure prediction on the alpha chain of human hemoglobin. Compare the predictions to those of NNSSP. 4. Determine whether the human transcription factor AP-1 (proto-oncogene C-JUN) has a coiled coil motif 5. Does the E. coli Lac repressor contain any recognizable folds? References Cuff J. A. and Barton G. J. (1999) Evaluation and improvement of multiple sequence methods for protein secondary structure prediction. PROTEINS: Structure, Function and Genetics 34:508-519. Erik L.L. Sonnhammer, Gunnar von Heijne, and Anders Krogh: A hidden Markov model for predicting transmembrane helices in protein sequences. In Proc. of Sixth Int. Conf. on Intelligent Systems for Molecular Biology, p 175-182 Ed J. Glasgow, T. Littlejohn, F. Major, R. Lathrop, D. Sankoff, and C. Sensen Menlo Park, CA: AAAI Press, 1998 (pdf download) Garnier J, Osguthorpe DJ, Robson B (1978) Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J Mol Biol 120(1):97-120 Accuracy of structure prediction methods Protein Structure Prediction Center Fold recognition Fold recognition links Tertiary structure prediction tools and structure databases SWISS-MODEL Modeller-4 SCOP Comprehensive lists of structure prediction sites can be found at: Index of resources Structure Prediction & Evaluation Protein Structure Prediction Some Other Structural Biology Databases and Servers around the world Network Protein Sequence Analysis Summary of protein structure Principles of Protein Structure, Comparative Protein Modelling and Visualisation Papers and essays on Tertiary structure prediction Pedestrian guide to analysing sequence databases Sisyphus and protein structure prediction A Guide to Structure Prediction (version 2)