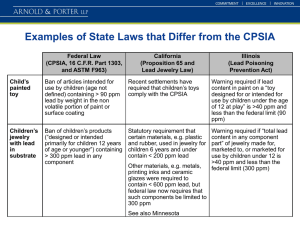
The Perfluorinated Alcohols (F5C6)(F3C)2COH and (F5C6)(F10C5
advertisement
(F3C)2COH and (F5C6)(F10C5")
The
Perfluorinated
Alcohols
(F5C6)(F3C)2COH
and
(F5C6)(F10C5)COH: Synthesis, Theoretical and Acidity Studies,
Spectroscopy and Structures in the Solid State and the Gas Phase
Nils Trapp,a Harald Scherer,a Stuart A. Hayes,b Raphael J.F. Berger,b Agnes
Kütt,c Norbert W. Mitzel,b Jaan Saamec and Ingo Krossing*a
a
Prof. Dr. Ingo Krossing, Dr. Nils Trapp, Dr. Harald Scherer, Albert-Ludwigs-Universität
Freiburg,
Institut
für
Anorganische
und
Analytische
Chemie
and
Freiburger
Materialforschungszentrum FMF, Albertstr. 21 D-79104 Freiburg (Germany), Fax:
(+49)7612036001, E-Mail: krossing@uni-freiburg.de (Preparation, spectroscopy, X-ray
diffraction, calculations).
b
Prof. Dr. Norbert W. Mitzel, Dr. Stuart Hayes, Dr. Raphael J. F. Berger, Universität
Bielefeld, Lehrstuhl für Anorganische Chemie und Strukturchemie, Universitätsstr. 25, D33615 Bielefeld (Germany) (Gas-phase electron diffraction).
c
Dr. Agnes Kütt, Jaan Saame, Institute of Chemistry, University of Tartu, 14a Ravila Street,
50411 Tartu (Estonia) (Acidity measurements).
Abstract
The syntheses of the perfluorinated alcohols (F5C6)(F3C)2COH (1) and (F5C6)(C5F10)COH (2)
are described. Both compounds were prepared in reasonable yields (1: 65%, 2: 85%) by
reacting the corresponding ketone with C6F5MgBr, followed by acidic work-up. The alcohols
were characterized by NMR, vibrational spectroscopy, single-crystal X-ray diffraction, acidity
measurements and gas-phase electron diffraction. A combination of appropriate 2D NMR
experiments allowed the unambiguous assignment of all signals in the
19
F spin systems, of
which that of 2 was especially complex. High acidity of the alcohols is indicated by acidity
measurements as well as the calculated gas phase acidities. It is also supported by the crystal
structure of 2, which exhibits only a single weak intermolecular hydrogen bridge with an
OO distance of 301 pm. This shows the low donor strength of the oxygen atom in the
compound, which is partly compensated through formation of two intramolecular CFH
contacts of 220 and 232 pm length to the proton not involved in the hydrogen bridge. The pKa
values in acetonitrile are 22.2 for 1 and 22.0 for 2; their calculated gas phase acidites are 1367
and 1343 kJ mol-1 (MP2/TZVPP level).
1
Introduction
Partly fluorinated and perfluorinated alcohols are used as specialty solvents, owing to their
low nucleophilicity,[1] high H-bond donor strength[2] and optical transparency. It was recently
shown, for example, that the oxidation of olefins by hydrogen peroxide is accelerated in
1,1,1,3,3,3-hexafluoro-2-propanol as a solvent by a factor of 105, if compared to 1,4dioxane.[3] Perfluorinated tertiary alcohols, such as nonafluoro-tert-butanol, share these
qualities and additionally exhibit exceptional chemical robustness, which can be exploited e.g.
in the synthesis of large and stable weakly coordinating anions.[4] However, the number of
easily available compounds in this class is small. Thus straightforward syntheses of alcohols
from common fluorinated precursors, which can be carried out in standard glass vessels, are
desirable as most laboratories are not equipped for direct fluorination techniques.
Perfluorinated tertiary alcohols can be prepared by reacting perfluorinated ketones with
perfluorinated organometallic reagents[5,
6]
and SbF5-catalyzed rearrangement of fluoro-
oxiranes.[7] The commercially available nonafluoro-tert-butanol can be obtained by CsFcatalyzed
disproportionation
of
fluoroacetone,[8]
by
halogen
exchange
from
(F3C)2(Cl3C)COH,[9] or even from the extremely toxic perfluoroisobutene.[10-12] More
recently, fluoroalkylsilanes such as F3CSi(CH3)3 were successfully used to transfer
fluoroalkyl groups to carbonyl compounds,[13, 14] anhydrides or activated esters of carboxylic
acids,[15] usually in the presence of fluoride as a catalyst.
Extensive studies on hydrogen bonding in fluorinated alcohols were published.[16-19] In
summary, the presence of electron-withdrawing groups adjacent to the hydroxyl function
makes the oxygen a weaker Lewis donor (weaker hydrogen bond acceptor), while the proton
is a stronger Lewis acceptor (stronger hydrogen bond donor). However, this higher Lewis
acidity does not compensate for the reduced basicity of the oxygen atom. As a result, there is
weaker intermolecular hydrogen bonding between the fluorinated alcohols themselves. Often
steric hindrance additionally reduces the extent of intermolecular hydrogen bonding. At the
same time strong interactions are observed between the fluorinated alcohol and a donor
molecule (such as ethers). For example, hexafluoroisopropanol forms a discrete complex with
tetrahydrofuran and other basic solvents.[2]
Results and discussion
(F5C6)(F3C)2COH (1) was prepared by a Grignard-type reaction originally described
elsewhere,[6] avoiding the erratically explosive C6F5Li,[20] with slight optimizations to increase
the total yield. C6F5MgBr was reacted with excess gaseous hexafluoroacetone in diethyl ether
2
and then hydrolyzed with aqueous HCl. The neat product can only be obtained by distillation
of the resulting ether adduct 1OEt2 from concentrated H2SO4 in 65 % overall yield.
(F5C6)(C5F10)COH (2) was formed in a similar reaction (100 g scale) from the Grignard
reagent and perfluorocyclohexanone, i.e. with no volatile reagents. The product is a colorless
powder, which was purified by sublimation (85 % overall yield).
OH
O
C6F5
CF3
CF3
or
C6F5MgBr
CF3
CF3
acidic work-up
or
C6F5
OH
CF2
F2C
CF2
CF2
F2C
O
F2C
F2C
C
F2
CF2
C
F2
Figure 1. Addition of the C6F5MgBr Grignard reagent to the fluorinated ketones, leading to
compounds 1 and 2 after acidic workup.
Both alcohols are soluble in ethers, toluene, chloroform, dichloromethane, acetonitrile,
fluorobenzene and 1,2-difluorobenzene. 2 is soluble in hexane. Other solvents were not tested.
Spectroscopic analyses
Infrared and Raman spectra of neat liquid 1 and solid 2 are in good agreement with calculated
bands (Figure 2, Table 1) and show the expected signals in the range of C-F and C-C bands.
The main O-H stretching modes were recorded at 3610 cm–1 for 1 and 3623 cm–1 for 2. Both
signals exhibit shoulders, indicating the existence of associated structures in their standard
state. In agreement with this observation, dimers were found in the crystal structure of 2 (see
below).
IR sim.
a)
IR sim.
b)
IR exp.
IR exp.
C6F5
OH
F2C
CF2
OH
C6F5
CF3
Raman exp.
F2C
CF3
Raman exp.
CF2
C
F2
Raman sim.
3800
3600
1600
1400
1200
1000
800
600
400
200
Raman sim.
3800
-1
wave number [cm
3600
1600
1400
1200
1000
-1
]
wave number [cm ]
3
800
600
400
200
Figure 2. Experimental (solid lines) and calculated (DFT-BP86/SV(P), dotted lines)
vibrational spectra of a) compound 1 and b) 2.
4
Table 1. IR and Raman bands of compounds 1 and 2, including assignment. Modes are described in terms of the closest comparable normal mode
where applicable and in general terms if the entire molecule is involved in the vibration.
1 exp. IR [cm–1][a]
422 (vw)
449 (vw)
477 (w)
540 (w), 552 (w)
646 (vw)
713 (mw), 733 (m)
744 (mw)
775 (vw)
804 (mw)
944 (m)
956 (m), 988 (m)
1008 (mw)
1110 (m), 1120 (m)
1140 (ms), 1152 (m)
1226 (s)
1275 (ms)
1313 (vw)
1368 (w)
1420 (w)
1498 (vs)
1533 (ms)
1617 (w)
1654 (mw)
3610 (mw), 3633 (w)
1 exp. Raman [cm–1][b]
217 (w)
235 (mw)
264 (m), 292 (vw), 306
(vw)
331 (mw)
347 (w)
356 (w), 379 (m)
424 (w)
449 (m)
480 (w)
499 (vs)
541 (vw), 553 (w)
588 (vs)
619 (w)
647 (vw)
714 (vw), 734 (vs)
775 (vw)
806 (m)
946 (vw)
956 (vw), 993 (w)
1014 (w)
1112 (vw), 1122 (vw)
1147 (w)
1238 (w)
1278 (vw)
1317 (vw)
1367 (vw)
1427 (w)
1492 (vw)
1535 (vw)
1656 (mw)
1 Assignment[c]
C-C, F-C-F
C6 out-of-plane
C-C, C-F
C-C, C-O-H twist
C-C, C-F
C-O-H twist
C-O-H twist
C-C
C6 out-of-plane
C6 out-of-plane
F-C-F
C6 stretch
C6 out-of-plane
C6 out-of-plane
C-C, F-C-F
C-C, F-C-F
C6 twist
C6 stretch
C-C
C-O-H bend
C-C
C-C, C-F
C-C, C-F
C-C, C-F
C-C-C
C-C-C, C-O-H
C6 stretch
C-O-H bend
C6 stretch
C6 twist
C6 stretch
C6 stretch
C6 stretch
O-H stretch
2 exp. IR [cm–1][a]
2 exp. Raman [cm–1][b]
154 (w)
200 (w)
280 (vw)
291 (vw)
315 (vw)
343 (mw)
376 (m)
412 (vw)
451 (mw)
476 (w)
513 (w)
584 (vw)
605 (vw)
622 (vw)
630 (w)
685 (vw)
730 (m)
787 (w)
849 (w)
917 (w), 947 (s)
992 (ms)
1017 (ms)
1034 (mw), 1105 (m)
1116 (m)
1137 (vw)
1155 (w), 1176 (m)
1183 (m)
1213 (s), 1239 (m)
1256 (w), 1307 (m)
1322 (m), 1345 (vw)
1383 (vw)
1405 (w), 1486 (vs)
1536 (m), 1616 (vw)
1651 (m)
3604 (mw), 3623 (m)
480 (m)
514 (m)
586 (ms)
630 (vw)
686 (vs)
919 (mw)
992 (ms)
1017 (ms)
1034 (mw), 1105 (m)
1116 (m)
1137 (vw)
1155 (w), 1176 (m)
1183 (m)
1213 (s), 1239 (m)
1256 (w), 1307 (m)
1322 (m), 1345 (vw)
1383 (vw)
1405 (w), 1486 (vs)
1536 (m), 1616 (vw)
1651 (m)
3604 (mw), 3623 (m)
2 Assignment[c]
arom. C6 out-of-plane
C-C, F-C-F
C-C, C-F, F-C-F
C-C, C-O-H bend
C-C, C-O-H twist
C-F, C-O-H bend
C-C, C-O-H twist
C-O-H twist
C-C, F-C-F
C-C
arom. C6 out-of-plane
aliph. C6 wagging
aliph. C6 stretch
C-C, F-C-F
arom. C6 out-of-plane
C-C
aliph. C6 stretch
C-C
arom. C6 twist
aliph. C6 twist
C-C, C-O-H bend
C-C, C-F
C-C, C-F
C-C, C-O-H bend
C-C, C-F
C-C
C-C, C-F
aliph. C6 wagging
aliph. C6 stretch
arom. C6 stretch
C-C, C-O-H bend
not calculated
arom. C6 stretch
arom. C6 stretch
arom. C6 stretch
O-H stretch
[a] ATR (ZnSe crystal, corrected for penetration depth); [b] FT-Raman (flame-sealed capillary); [c] based on BP86/SV(P) frequency calculation.
5
NMR spectroscopic characterization
The
19
F-NMR spectrum of 1, recorded at room temperature in CDCl3, shows the typical
chemical shifts to be expected for pentafluorophenyl groups at –160.2 ppm for the signal of
the meta-fluorine atoms and –148.9 ppm for the fluorine atom in para-position. The orthofluorine atoms are not chemically equivalent and display two broad resonances (FWHM ≈ 300
Hz) at –138.4 ppm and –132.8 ppm. This indicates a strongly hindered rotation of the
pentafluorophenyl ring also responsible for the broadening of the signal of the meta-fluorine
atoms. The resonance of the two CF3 groups is observed at –76.2 ppm. All CF3 fluorine atoms
are chemically equivalent, but there are two slightly different 5J(19F,19F) coupling constants to
the ortho-fluorine atoms. In the 1H-NMR spectrum one broad signal without observable
splitting is found at 4.13 ppm. Its linewidth (FWHM = 8 Hz) rules out 19F,1H-couplings bigger
than 4 Hz. The carbon chemical shifts could be easily assigned by 13C{1H}- and 2D-13C,19Fcorrelation spectra.
NMR spectra of 2 were recorded in toluene-d8 solutions at room temperature. The
19
F NMR
spectrum displays nine signals due to the fact that the fluorine atoms of the CF2 groups of the
cyclohexyl ring are not chemically equivalent. The spin systems in the aromatic and the
aliphatic part of the molecule are of higher order, resulting in complex splitting patterns. For
the resonances of the CF2 groups these patterns are dominated by the large 2J(19F,19F)
couplings with absolute values larger than 270 Hz, making them easy to identify. The 13C,19Fcorrelation spectrum (Figure 3A) allows to determine which fluorine resonances belong to the
same CF2 group. The
19
F signals at –113.6 ppm and –131.2 ppm correlate to the same
13
F resonances at –117.7 ppm and –135.8 ppm to the
13
F signals at –121.3 ppm and –140.7 ppm to the
13
resonance at 112.0 ppm (2), the
19
signal at 109.0 ppm (3) and the
19
C
C
C
resonance at 108.1 ppm (4), respectively. Because of their intensities, which are half of the
integrals of the other aliphatic fluorine resonances, the signals at –121.3 ppm and –140.7 ppm
are assigned to the CF2 group (4) of the cyclohexyl ring.
Typical values of the
13
C and
19
F chemical shifts are detected for the pentafluorophenyl
group. The resonance of the meta-fluorine atoms is found at –159.0 ppm, of the para-fluorine
atom at –146.7 ppm and of the ortho-fluorine atoms at –132.9 ppm. The signal of the orthofluorine atoms shows a significant line broadening, indicating a hindered rotation of the
pentafluorophenyl ring. The linewidth of this signal precludes resolving of the splitting
pattern, except for a triplet splitting of 53 Hz that cannot be explained solely by the spin
system of the pentafluorophenyl group and must thus be attributed to an additional coupling
to two fluorine atoms of the cyclohexyl ring. The absolute magnitude of this formal
6
5
J(19F,19F) coupling strongly indicates a major contribution of through-space interactions of
the coupling fluorine atoms. Given the structure of the molecule, the likeliest coupling
partners responsible for this triplet splitting of the ortho-fluorine resonances are the axial
fluorine atoms of the CF2 groups 2.
Figure 3. 2D NMR correlation spectra of 2 in toluene-d8 at RT (Avance II+ 400 MHz). A:
13
C,19F HSQC (1/(2J) = 1,25 ms); B: 19F NOESY (mixing time: 0.8 s, negative intensities in
solid black); C: 19F,1H COSY; lower left: Labelling of the atoms in the cyclohexyl ring of 2 as
used in the NMR discussion (R = C6F5).
7
In a 19F,19F COSY spectrum (see supplementary information) the signal of the ortho-fluorine
atoms correlates to the resonances at –113.6 ppm and –131.2 ppm, which must hence be
assigned to the CF2 groups 2, leaving the fluorine signals at –117.7 ppm and –135.8 ppm for
the CF2 groups 3 of the cyclohexyl ring. Nevertheless, there are cross-peaks of the orthofluorine resonances to both the fluorine signals of the CF2 groups 2. An analysis of the
splitting patterns of the signals in the 1D
19
F-NMR spectrum shows clearly that the 53 Hz
coupling is found in the resonance at –113.6 ppm, which is therefore assigned to the fluorine
atoms in axial position of these CF2 groups.
An independent confirmation of this assignment together with additional information about
the orientation of the aliphatic fluorine atoms can be obtained by homonuclear
19
F NOE
measurements, which are not yet commonly used in 19F-NMR.[21] In fact, to our knowledge it
is only the fifth example in literature where homonuclear
tool.
[22-25]
In the
19
19
F NOEs are used as an analytical
F,19F NOESY (Figure 3B) only one cross-peak is found between the
resonance at –113.6 ppm (2a) and signals of the neighbouring CF2 group 3, whereas for the
associated signal at –131.2 ppm (2e) there are cross-peaks to both resonances of this CF2
group. This unambiguously proves that the signal at –113.6 ppm must be assigned to the
fluorine atom in axial position of the CF2 group 2 and the signal at –131.2 ppm to the
corresponding fluorine atom in equatorial position. The one CF2 fluorine signal of CF2 group
3 that shows a cross-peak to the signal at –113.6 ppm and consequently belongs to the
fluorine atom in the equatorial position of this CF2 group observed at –135.8 ppm (3e). The
resonance at –117.7 ppm (3a) must hence be the signal of the fluorine atom in axial position
of this CF2 group. Finally there is a NOE cross peak between the signal at –113.6 ppm (2a)
and the resonance at –121.3 ppm (4a) of CF2 group 4, which is assigned to the fluorine atom
in axial position, leaving the signal at –140.7 ppm (4e) for the corresponding equatorial
fluorine atom. All other NOE cross-peaks are in accordance to this assignment. By this
means, all aliphatic fluorine resonances and the positions of the fluorine atoms in the
cyclohexyl ring are clearly assignable. It should be noted that as a result, the signals of the
fluorine atoms in axial and equatorial positions can be distinguished by their chemical shift,
which is about 18 ppm to higher field for the resonances of the fluorine atoms in equatorial
position. Unfortunately the cross-peaks of the resonance at –113.6 ppm (2a) to the orthofluorine signal are dispersive, which indicates magnetization transfer via spin-spin coupling
for these cross-peaks, superimposing potential NOE.
The 1H NMR spectrum shows the OH proton signal as a triplet at 3.14 ppm. In the
19
F,1H
COSY-NMR spectrum (Figure 3C) cross-peaks of this proton signal to the ortho-fluorine
8
resonances and to the fluorine signals of 2a, 2e and 3a are detected. The cross-peak to the
ortho-fluorine signal is the most intense, and unlike the others shows no triplet splitting in the
proton dimension but a doublet with the middle signal of the triplet missing. Taking the
mechanism of magnetization transfer for COSY-type experiments into account, this proves
that the
19
F,1H-coupling constant of 5.3 Hz is the active coupling for this cross-peak and
hence is the coupling between the proton and the ortho-fluorine atoms. For formal 5J(19F,1H)
coupling constants of this order of magnitude in such a structure, a decisive contribution of
through-space coupling is most likely.[26]
Thus a conformer with the proton pointing into the direction of the perfluorophenyl group
should be favored in solution as it is also found for the calculated molecular structures of 2
discussed below. A
19
F,1H HOESY (see supplementary information) contributes additional
confirmation for this interpretation. However, this spectrum also demonstrates that
conformers with the proton directed to the cyclohexyl ring exist as well.
Gas-phase electron diffraction investigation of 1
GED (gas-phase electron diffraction) data were recorded for 1 and its molecular structure was
refined making use of the SARACEN method[27] of structural refinement. An important
structural feature of this compound is the possibility of intra-molecular hydrogen bonding,
however, the positions of hydrogen atoms are usually not well-determined by GED due to the
relatively low electron-scattering cross-section of the hydrogen atom. On the other hand, as
there are no elements heavier than fluorine present in 1, it was possible that the diffraction
from hydrogen might not be completely lost, thus, models with and without the hydrogen
atom were tested. Despite the lack of any heavy atoms that would have dominated the
electron scattering, the two models gave an equally good fit of the theoretical and
experimental intensities, with almost identical R-factors and refined heavy-atom parameters.
In the model containing the hydrogen atom, the uncertainties on the parameters describing the
hydrogen atom positions were equal to the applied restraints, reinforcing the conclusion that
there is no information in the experimental data in this regard.
The refined geometric molecular parameters, for which structural information could be
obtained and the refined values are displayed in Table 2, whilst the molecular-intensity and
radial-distribution curves for 1 are shown in Figures 6 and 7, respectively. The radialdistribution curve (RDC, Figure 5) exhibits five distinct peaks (the first of which comprises
all of the bonded distances), two shoulders and an elongated tail. Each of these features can be
interpreted as having the potential to provide some structural information, whilst the
9
remaining features, such as the peak shapes and relative heights, may contain further
information that will generally be less certain. In any case, the molecule is too large for a full
structural refinement based on the GED data alone, thus a combination of constraints and
restraints was applied, along the lines of the SARACEN method[27, 28] of structural refinement.
The constraints used were based on the results of ab initio calculations and are in line with the
local symmetry suggested by solution NMR spectroscopy, namely pseudo-C2v symmetry for
the perfluorophenyl group (the deviation from this symmetry is due to independent placing of
the ortho-fluorine atoms), and assumption of a single internal F–C–F angle for the two CF3
groups. The molecular structure (Figure 6) was thus defined in terms of 34 independent
parameters (a full description of the model and the independent parameters used is provided
in Supplementary Information). Of these, only six parameters were refined without being
directly restrained and a further three parameters refined with only loose restraints, yielding
uncertainties significantly smaller than the corresponding restraint uncertainty, therefore
indicating the influence of the experimental data in the refined structure. However, due to the
similarity of the bonded distances, as evident from the radial distribution curve, the aromatic
C–C, the C–O and the C–F distances are correlated, and the differences between the aromatic
C–C distance, and the C–O and C–F distances were restrained to calculated values, as
summarized in Table 2. In addition to these parameters, the C–C–O angle could also be
refined without a restraint, converging to 115.8(8)° or 114.2(8)° depending on whether the
respective model for vibrational motion adopted was rg or rh1. Only the first of these values is
significantly larger than the calculated value of 112.8°, however, the disagreement between
the two structure types gives a more faithful estimate of the parameter uncertainty than just
the standard deviation obtained from the least-squares refinement (which assumes a particular
model). In the end, this parameter was restrained to the calculated value, as shown in Table 2,
without any detrimental effect on the quality of the refinement.
Table 2. Ten selected (of 34) independent parameters (p) for which information was obtained
in the GED study and dependent parameters (d) that were restrained during the GED
refinement, as well as restraints and their uncertainties. The atom numbering is shown in
Figure 6.
Independent parameters
GED (rg)
GED (rh1) restraint
MP2/TZVPP (re)a
p1
r C–C average (aromatic)
1.404(2)
1.400(2)
1.391
p3
r C–C average (aliphatic)
1.556(5)
1.549(5)
1.538
p6
r C–O
1.409(5)
1.409(5)
1.398
p8
r C–F average
1.339(1)
1.340(1)
1.332
p21
C(1)–C(12)–O
113.2(4)
113.4(4)
10
112.8(5)
112.8
p23
C(4)–C–C–O
31.0(15)
29.8(19)
p25
C(1)–C(12)–C average
110.6(4)
110.2(4)
111.0
p29
F–C–F
107.5(2)
107.2(2)
108.0
p32
C(1)–C–C–F(16)
86.2(9)
85.8(8)
84.6(50)
84.6
p33
C(1)–C–C–F(20)
65.4(23)
66.7(22)
67.3(50)
67.3
31.5(100)
31.5
Dependent parameters
d1
r
C–O
minus
C–C 0.006(5)
0.009(5)
0.007(5)
0.007
d2
r C–C aromatic minus C–F 0.065(3)
0.060(4)
0.059(5)
0.059
R-Factor
6.60 %
6.52 %
–
a
r
(
o
p
m
1
a
t
m
i
i
c
n
u
(
s
p
6
p
8
m
)
i
n
u
s
p
1
)
Figure 4. Experimental, theoretical and difference (experimental minus theoretical)
molecular-scattering intensities.
11
Figure 5. Experimental and difference (experimental minus theoretical) radial-distribution
curves. Molecular-scattering intensities were multiplied by sexp[(210–6 s2)/(ZF–fF)(ZO–fO)]
prior to Fourier transformation.
(a) 0.0 kJ mol–1
(b) 1.7 kJ mol–1
Figure 6. Molecular geometries of 1, calculated to be minima on the potential energy surface
and their relative electronic energies (MP2/TZVPP). Atom numbering is as used in the GED
refinement.
Theoretical calculations suggest two possible conformers, as evident by local minima on the
potential energy surface and that are shown in Figure 6: One with the hydrogen atom in the
cis conformation with respect to the perfluorinated phenyl moiety (Fig. 9a) and another in the
trans position (Fig. 9b). Calculations at both the MP2 and B3LYP levels of theory predict the
12
cis conformer to have the lowest energy, but the calculated energy difference is smaller than
can be accurately calculated and, as mentioned above, the two conformers could not be
distinguished by GED. However, as can be seen from Table 2, the calculated results are in
good agreement with those from GED, thus it seems that the MP2 method performs well for
this type of chemical system. Full details of the structural model, refined parameter values
(rh1), molecular geometries for both the rh1 and rg structure types, data analysis parameters,
correlation matrix and up-hill curves are provided as Supplementary Information.
Crystal structure analysis of 2 and results from computations
Compound 2 crystallized at room temperature from a hexane solution in the form of colorless
needles. The space group was determined as monoclinic P21/c with a = 12.05 Å, b = 15.16 Å,
c = 15.99 Å, β = 113.9° and Z = 8.
Figure 7. Asymmetric unit of the crystal structure of 2. Thermal ellipsoids drawn at 50%
probability level. Distances are given in pm, libration-corrected distances are indicated by an
asterisk: O1-O2 301.2(3) pm, O1-H1 93(4)*, O2-H2 88(3)*, O1-C 141.7(1), O2-C 140.0(1),
O1-H2-O2 166.3(2)°.
The asymmetric unit contains a dimer of 2 (Figure 7) connected by an asymmetric hydrogen
bridge. All contacts fall into the characteristic range for weak O-HO bridges[29,
30]
with
H2O1: 223.1(1) pm, H2-O2: 88(3) pm (corrected for libration), O1O2: 301.2(1) pm and
an O-H-O angle of 166(3)°. The H atom not involved in the H bonding points away from the
hydrogen bridge with a H-O-H angle of 119.1(1)° (both hydrogen atoms were found on the
Fourier map and refined without positional constraints). This bonding scenario is in
13
agreement with the considerations from earlier studies, already described above.
Perfluorinated alcohols tend to be poor donors for intermolecular H bonding due to the
presence of electron-withdrawing groups near the oxygen atom. In the absence of a donor and
in combination with the high steric demand of the C12F15 moiety, this leads to the formation
of only a single weak hydrogen bridge. Correspondingly, in the molecule which acts as
hydrogen donor, the C6F5 group is rotated by 81.7° with respect to the C6F10 group, while in
the other both groups are orthogonal with an angle of 89.7°.
Expectedly, in the MP2/TZVPP calculation of the monomer without any hydrogen bonding,
the angle lies between both values with 85.0°. The calculated contact distances between the
fluorine atoms in the ortho position of the C6F5 ring and the 2-position of the C6F10 ring are
243 and 255 pm, which is in good accordance with the crystal structure (243 to 263 pm). The
proton not involved in the intermolecular hydrogen bond additionally exhibits two rather short
HF contacts at 220 pm (with the fluorine atom in ortho position of C6F5, OF: 250 pm) and
232 pm (with a fluorine atom in C6F10, OF: 273 pm) in the crystal structure. These
interactions are comparable to the HF interactions in 1 (HF (calc., MP2/TZVPP): 246 and
184 pm, OF (calc.) 271 and 260 pm) and in good agreement with the MP2/TZVPP
minimum geometry (which features OF distances of 250 and 269 pm, respectively). Apart
from the global minimum, a Cs symmetric local minimum only 13 kJ mol–1 higher in energy
was found (Figure 8).
Figure 8. H-F contacts and F-F contacts in a) the C1 global minimum geometry of 2; b)
another local minimum structure with Cs symmetry. Geometries were calculated at the
MP2/TZVPP level (distances given in pm).
14
This is in good agreement with the NMR spectroscopic results. As discussed above, these
indicate a dynamic system in solution, in which the global minimum with the H atom pointing
in the direction of the pentafluorophenyl group is slightly favoured.
Another crystal structure (2a) could be recorded, in which 2 co-crystallized with nbutylammonium bromide and one water molecule. The crystal quality was very poor, hence
the structure is regarded as preliminary and shall not be discussed in much detail. However, it
exhibits a very short hydrogen bridge between the hydroxyl group of 2 and the water
molecule (OO distance: 254 pm, cf. 250-260 pm in H9O4+). This is another clear indication
of the low basicity of the alcohol and underlines its potential in further syntheses. More
details of this structure are deposited with the supplementary information.
Acidities of the perfluorinated alcohols
Table 3 shows calculated and experimental acidity values of compounds 1 and 2 in
comparison with the acidic perfluorinated alcohols nonafluoro-tert-butanol 3 and perfluoro-1adamantanol C10F15OH 4. For experimental pKa values in acetonitrile of a wider range of
acids, including non-fluorinated species, see Refs.[31] and [32].
Table 3. Experimental pKa and logKAHA values in acetonitrile and experimental and calculated
(MP2/TZVPP) gas phase acidities (GA, kJ mol–1) of fluorinated alcohols including
compounds 1 and 2.
Compound
pKa (MeCN)
logKAHA
GA (exp.)
GA (calc.)
(F5C6)(F3C)2COH 1
22.15
4.7
-
1367
(F5C6)(F10C5)COH 2
22.00
4.7
-
1343
(F3C)3COH 3
20.55[31]
4.8[31]
1355[33]
1366
C10F15OH 4
18.25
4.8
1321[34]
1328
The acidities of 1 - 4 are governed on one hand by the intrinsic properties of the compounds
and on the other hand by solvent effects. The gas-phase acidities are influenced by the
intrinsic properties only. The solvent acidities (pKa values) are influenced both by the intrinsic
properties and by the solvent effects.
The following intrinsic properties are important:
(1) the inductive effect of the -fluorine atoms;
15
(2) release of steric strain via shortening of the C-O bond upon deprotonation and
simultaneous elongation of the C-C bond (see ref. [35] for examples of the high importance
of steric strain release on acidity of strained polyfluorinated alcohols);
(3) possible weak intramolecular hydrogen bond in the neutral molecules 1 and 2;
(4) steric shielding of the anionic centre in the anions and the –OH group in the neutrals;
(5) polarizability of the fluorocarbon moiety of the alcohol;
The first two properties are the most important both in the gas phase and in solution. The
number of -fluorine atoms is 6 in 1, 4 in 2, 9 in (F3C)3COH 3 and 6 in perfluoroadamantanol
4. Release of steric strain is an acidifying factor in perfluoroadamantanol, but probably not in
the other three alcohols (see ref. [34]).
In the gas phase, where polarizability and size are very important for the higher acidity 2 is a
markedly stronger acid than 1 (by 24 kJ mol-1) and the rather bulky perfluoroadamantanol 4 is
the strongest (15 kJ mol-1 from 2) of all measured alcohols. Even though 3 has the largest
number of -fluorine atoms (and it is clearly stronger than 1 and 2 in acetonitrile), in the gas
phase it is significantly weaker than 2 and comparable to 1. There is also a possibility of a
weak intramolecular hydrogen bond in compounds 1 and 2 (Figure 6).
In acetonitrile, in addition to the intrinsic effects solvation of the anions is important. The
anions of all four alcohols have localized charges and even with a poor H-bonding donor such
as MeCN, there is some solvent effect. Also, steric shielding of the anionic centre comes into
play. Looking at the geometries of the anions, the –O– centre seems to be most shielded in 2
and least shielded in perfluoroadamantanol 4. Taking into account these and the above
considerations, it appears logical that in MeCN perfluoroadamantanol 4 is the most acidic of
the three compounds, followed by (F3C)3COH 3, and 1 and 2 are the weakest. All of
measured compounds have high homoconjugation constants in acetonitrile. This is an
indication of unfavorable solvation of the perfluoroalcoholate anions in acetonitrile, so that
the anions are stabilized via hydrogen bonding with the corresponding neutral compounds
(which are also very strong hydrogen bond donors) present in the solution.
Conclusions
The perfluorinated alcohols (F5C6)(F3C)2COH (1) and (F5C6)(C5F10)COH (2) were
synthesized and fully characterized; compound 2 is available in a convenient 100 g scale
16
reaction and easily purified by sublimation. 2 exhibits a weak intermolecular OH bridge and
two short intramolecular CFH contacts, indicative of the low donor strength and thus high
acidity of the compound. These interactions complicated the already non-trivial NMR studies
even further; however the spectra could still be fully assigned and interpreted using a
combination of (2D-)NMR experiments, crystal structure data and computations. Both
compounds, as well as their metal alkoxides (which will be discussed in detail in an upcoming
publication), are promising precursors for further chemistry with perfluorinated ligands. There
are especially interesting prospects for crystallography, as the number of rather rigid, stable
perfluorinated ligands such as in 2 is still limited. While not quite as acidic as e.g.
(F3C)3COH, the examined alcohols are still very suitable as ligands in stable and weakly
coordinating anions exhibiting no crystallographic disorder, as will be shown in upcoming
publications.
Experimental
General
Bromopentafluorobenzene was purchased from ChemPur in 99 % purity and purified by
distillation after stirring over P2O5. Hexafluoroacetone and perfluorocyclohexanone were
purchased from P&M Invest. The gaseous hexafluoroacetone was used without further
purification, whereas the ketone was stirred over P2O5, then condensed. Diethyl ether was
dried in a Grubbs apparatus and degassed prior to use. Raman spectra were recorded using a
Bruker Vertex 70 spectrometer with a RAM II FT-Raman module (excitation wavelength
1064 nm, 20 mW laser power). ZnSe-ATR (attenuated total reflection) IR spectra were
recorded on a Nicolet Magna-IR 760 FT-IR spectrometer. NMR spectra were recorded in
purified deuterated solvents on a Bruker Avance 400 MHz spectrometer. Melting points were
determined with a Krüss KSPS 1000 digital melting point analyzer. Crystal structures were
determined at 110 K on a Rigaku R-Axis SPIDER diffractometer using a sealed-tube Mo
source. Gas-phase electron scattering was recorded at room temperature on reusable Fuji
imaging plates with a Balzers KD-G 2 Gas-Eldigraph, equipped with a Staib Instruments
electron source (50 kV/200 nA).[36] Calculations were carried out with the TURBOMOLE
program package.[37] Calculated compounds were investigated with DFT-BP86,[38, 39] as well
as second-order Møller-Plesset (MP2[40, 41]) perturbation calculations. SV(P) and TZVPP[42]
basis sets were used and the RI approximation[40,
41]
) was applied. Vibrational frequencies
were calculated analytically with the AOFORCE module[43] at the BP86/SV(P) level, and the
results were used to determine the zero point vibrational energy (ZPE) and to verify that the
17
geometry optimization resulted in a local minimum on the potential energy hypersurface with
no imaginary frequencies.
Acidity measurements
The pKa measurement procedure as well as the definitions of Ka, pKa and KAHA are the same
as in ref [31].
Synthesis of 1,1,1,3,3,3-hexafluoro-2-pentafluorophenyl-ipropanol (1)
Mg filings (2.50 g, 93.1 mmol) were weighed into a 250 ml round bottom flask fitted with a
dropping funnel and reflux condenser (cooled to 0 °C). Diethyl ether (150 ml) was added and
the flask kept at 0 °C. C6F5Br (11.1 ml, 89.0 mmol) was added dropwise over 30 minutes. The
mixture turned turbid, then dark brown. The reaction was then stirred for 16 h at RT. The
condenser and flask were cooled to –60 °C and gaseous hexafluoroacetone (109.8 mmol) was
condensed into the flask. The flask was allowed to reach RT and the mixture was stirred for a
further 12 h. The solvent was then removed under reduced pressure and the residue
hydrolyzed with excess aqueous HCl (32 %, 30 ml). After stirring for 2 hours, HCl and water
were removed by condensation under reduced pressure. MgBrCl was removed by filtration
and washed twice with diethyl ether (50 ml). The filtrate was separated from the solvent by
fractioned distillation. To remove residual diethyl ether, the distillate was again distilled from
concentrated H2SO4 (b.p. 162 °C) to afford 19.3 g (65 %) of clear colorless 1. 1H NMR (400
MHz, CDCl3, RT): δ = 4.13 ppm (s). 13C[1H] NMR (50 MHz, CDCl3, RT): δ = 79.1 (m, 2JC,F
= 33 Hz), 105.4 (m, 2JC,F = 11 Hz), 121.6 (q, 1JC1JC,F = 288 Hz), 138.3 (m, 1JC,F = 253 Hz,
2
JC,F = 12.4 Hz, 2JC,F = 18.7 Hz), 142.6 (m, 1JC,F = 259 Hz, 2JC,F = 13 Hz, 3JC,F = 6 Hz), 146.3
(d, 1JC,F = 259 Hz) ppm.
19
F NMR (377 MHz, CDCl3, RT): δ = –76.2 (m, 5JF,F = 15 Hz), –
132.8 (b), –138.4 (b), –148.9 (m, 3JF,F = 23 Hz, 4JF,F = 7 Hz), –160.2 (b) ppm. IR (ZnSe
ATR): υ = 422 (vw), 449 (vw), 477 (w), 540 (w), 552 (w), 646 (vw), 713 (mw), 733 (m), 744
(mw), 775 (vw), 804 (mw), 944 (m), 956 (m), 988 (m), 1008 (mw), 1110 (m), 1120 (m), 1140
(ms), 1152 (m), 1226 (s), 1275 (ms), 1368 (w), 1420 (w), 1498 (vs), 1533 (ms), 1617 (w),
1654 (mw), 3610 (mw) cm–1.
Synthesis of 2,2,3,3,4,4,5,5,6,6-decafluoro-1-pentafluorophenyl-cyclohexanol (2)
Mg filings (3.00 g, 123 mmol) were weighed into a 500 ml round bottom flask fitted with a
dropping funnel and reflux condenser (cooled to 15 °C). Diethyl ether (200 ml) was added
and the flask kept at 0 °C. C6F5Br (15.0 ml, 120 mmol) was added dropwise over 30 minutes.
18
The mixture turned turbid, then dark brown. The reaction was then stirred for 16 h at RT.
Perfluorocyclohexanone (19.0 ml, 117 mmol), dissolved in diethyl ether (20 ml), was added
dropwise over 30 minutes. The reaction mixture was refluxed for 4 h and cooled to RT.
Aqueous HCl (32 %, 30 ml) was added and the mixture stirred for 2 h. HCl and water were
removed by condensation under reduced pressure and the product was isolated by sublimation
(10–2 mbar, 50 °C) to afford 53.6 g (85 %) of solid colorless 2 (m.p. = 52 °C). 1H NMR (400
MHz, CDCl3, RT): δ = 4.20 (t, JH,F = 6.2 Hz) ppm. 13C[1H] NMR (101 MHz, toluene-D8): δ =
78.9 (m, 2JC,F = 26 Hz), 108.1 (b), 109.0 (b), 112.0 (b), 138.1 (d, 1JC,F = 247 Hz), 142.2 (d,
1
JC,F = 260), 146.4 (d, 1JC,F = 258 Hz). 19F NMR (377 MHz, toluene-D8): δ = –113.6 (m, 2JF,F
= 283 Hz, 5JF,F = 53 Hz), –117.7 (m, 2JF,F = 274 Hz), –121.3 (m, 2JF,F = 284 Hz), –131.2 (m,
2
JF,F = 283 Hz), –132.9 (m, 5JF,F = 53 Hz), –135.8 (m, 2JF,F = 274 Hz), –140.7 (m, 2JF,F = 284
Hz), –146.7 (m, 3JF,F = 22 Hz, 4JF,F = 7 Hz), –159.0 (m) ppm. IR (Diamond ATR): υ = 476
(w), 513 (w), 584 (vw), 605 (vw), 622 (vw), 630 (w), 685 (vw), 730 (m), 787 (w), 849 (w),
917 (w), 947 (s), 992 (ms), 1017 (ms), 1034 (mw), 1105 (m), 1116 (m), 1137 (vw), 1155 (w),
1176 (m), 1183 (m), 1213 (s), 1239 (m), 1256 (w), 1307 (m), 1322 (m), 1345 (vw), 1383
(vw), 1405 (w), 1486 (vs), 1536 (m), 1616 (vw), 1651 (m), 3623 (m) cm –1. FT-Raman: υ =
154 (w), 200 (w), 280 (vw), 291 (vw), 315 (vw), 343 (mw), 376 (m), 412 (vw), 451 (mw),
480 (m), 514 (m), 586 (ms), 630 (vw), 686 (vs), 919 (mw), 1087 (vw), 1139 (w), 1236 (w),
1259 (w), 1299 (w), 1322 (w), 1405 (w), 1652 (mw) cm–1. The crystal structure of 2 was
submitted to the Cambridge Structural Database, from where it can be retrieved free of charge
under the reference number CCDC 800116.
Acknowledgements
The authors gratefully acknowledge financial support from the DFG, FCI and the University
of Freiburg as well as from the Estonian Science Foundation (grant No 7374). We also thank
Prof. Ivo Leito for fruitful discussions and corrections and Prof. Gerhard Hägele for helpful
advice concerning the NMR studies.
Notes and references
[1]
T. W. Bentley and P. v. R. Schleyer, Adv. Phys. Org. Chem. 1977, 14, 1.
[2]
W. J. Middleton and R. V. Lindsey, Jr., J. Am. Chem. Soc. 1964, 86, 4948.
[3]
A. Berkessel and J. A. Adrio, J. Am. Chem. Soc. 2006, 128, 13412.
[4]
I. Krossing, Chem. Eur. J. 2001, 7, 490.
[5]
G. J. Moore, C. F. Smith and C. Tamborski, J. Fluorine Chem. 1975, 5, 77.
19
[6]
C. Tamborski, W. H. Burton and L. W. Breed, J. Org. Chem. 1966, 31, 4229.
[7]
R. J. DePasquale, K. B. Baucom and J. R. Patton, Tetrahedron Lett. 1974, 13, 1111.
[8]
V. Weinmayr, Patent: U.S. 3317616, 1967.
[9]
R. Filler and R. M. Schure, J. Org. Chem. 1967, 32, 1217.
[10]
B. L. Dyatkin, E. P. Mochalina, R. A. Bekker, S. R. Sterlin and I. L. Knunyants,
Tetrahedron 1967, 23, 4291.
[11]
I. L. Knunyants, B. L. Dyatkin, A. V. Fokin and V. A. Komarov, Izv. Akad. Nauk
SSSR, Ser. Khim. 1964, 1425.
[12]
I. L. Knunyants and B. L. Dyatkin, Izv. Akad. Nauk SSSR, Ser. Khim. 1964, 923.
[13]
S. P. Kotun, J. D. O. Anderson and D. D. Des Marteau, J. Org. Chem. 1992, 57, 1124.
[14]
N. R. Patel and R. L. Kirchmeier, Inorg. Chem. 1992, 31, 2537.
[15]
L. A. Babadzhanova, N. V. Kirij, Y. L. Yagupolskii, W. Tyrra and D. Naumann,
Tetrahedron 2005, 61, 1813.
[16]
A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc. 2006,
128, 8421.
[17]
K. F. Purcell, J. A. Stikeleather and S. D. Brunk, J. Mol. Spectr. 1969, 32, 202.
[18]
R. M. Guidry and R. S. Drago, J. Am. Chem. Soc. 1973, 95, 759.
[19]
O. Schrems, H. M. Oberhoffer and W. A. P. Luck, J. Phys. Chem. 1984, 88, 4335.
[20]
P. L. Coe, R. Stephens and J. C. Tatlow, J. Chem. Soc. 1962, 3227.
[21]
R. J. Battiste and A. Newmark, Prog. Nuc. Mag. Res. Spec. 2006, 48, 1.
[22]
M. F. Mahon, M. K. Whittlesey and P. T. Wood, Organomet. 1999, 18, 4068.
[23]
J. L. Battiste, N. Jing and R. A. Newmark, J. Fluorine Chem. 2004, 125, 1331.
[24]
G. W. Buchanan, E. Munteanu, B. A. Dawson and D. Hodgson, Magn. Reson. Chem.
2005, 43, 528.
[25]
W. Tyrra, H. Scherer, L. A. Babadzhanova, N. V. Kirij, Y. L. Yagupolskii, D.
Naumann and I. Pantenburg, Helv. Chim. Acta 2008, 91, 97.
[26]
L. Hennig, K. Ayala-Leon, J. Angulo-Cornejo, R. Richter and L. Beyer, J. Fluorine
Chem. 2009, 130, 453.
[27]
N. W. Mitzel and D. W. H. Rankin, J. Chem. Soc., Dalton Trans. 2003, 3650.
[28]
A. J. Blake, P. T. Brain, H. McNab, J. Miller, C. A. Morrison, S. Parsons, D. W. H.
Rankin, H. E. Robertson and B. A. Smart, J. Phys. Chem. 1996, 100, 12280.
[29]
G. A. Jeffrey, Cryst. Rev. 2003, 9, 135.
[30]
T. Steiner and W. Saenger, Acta Cryst. B 1992, B48, 819.
20
[31]
A. Kütt, I. Leito, I. Kaljurand, L. Sooväli, V. M. Vlasov, L. M. Yagupolskii and I. A.
Koppel, J. Org. Chem. 2006, 71, 2829.
[32]
F. Eckert, I. Leito, I. Kaljurand, A. Kütt, A. Klamt and M. Diedenhofen, J. Comp.
Chem. 2009, 30, 799.
[33]
R. W. Taft, I. A. Koppel, R. D. Topsom and F. Anvia, J. Am. Chem. Soc. 1990, 112,
2047.
[34]
R. Herrero, J. Z. Davalos, J.-L. M. Abboud, I. Alkorta, I. A. Koppel, T. Sonoda and
M. Mishima, Int. J. Mass Spectrom. 2007, 267, 302.
[35]
L. Lipping, I. Koppel, I. A. Koppel, A. Kolomeitsev, G.-V. Röschenthaler and I. Leito,
J. Org. Chem. 2010, 75, 6436.
[36]
R. J. F. Berger, M. Hoffmann, S. A. Hayes and N. W. Mitzel, Z. Naturforsch. 2009,
64b, 1259.
[37]
R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett. 1989, 162,
165.
[38]
J. P. Perdew, K. Burke and Y. Wang, Phys. Rev. B 1996, 54, 16533.
[39]
A. D. Becke, Phys. Rev. A 1988, 38, 3098.
[40]
F. Weigend and M. Häser, Theor. Chem. Acc. 1997, 97, 331.
[41]
F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett. 1998, 294, 143.
[42]
A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys. 1994, 100, 5829.
[43]
P. Deglmann, F. Furche and R. Ahlrichs, Chem. Phys. Lett. 2002, 362, 511.
21