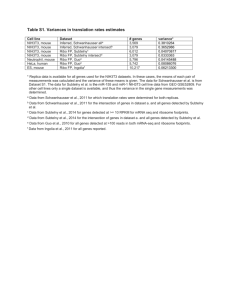
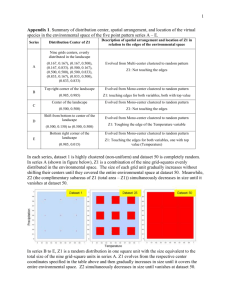
supplementary material
advertisement

Continuous Hidden Process Model for Time
Series Expression Experiments
Supplementary Materials
Yanxin Shi, Michael Klutstein, Itamar Simon, Tom Mitchell and Ziv Bar-Joseph
1.
Detailed derivations
Inference:
Inference on CHPM is a special case of Kalman filter where the transition matrix is
fixed to identity matrix and the covariance matrices for transition model and emission
model are diagonal. Here we only state the final results without proof. Please see e.g.
[Jor02, Mur02] for derivations.
Forward pass
Let Pt|t and Vt|t denote the mean and variance of the Pt given G1:t, respectively, where
G1:t is the observed genes’ expression levels from time point 1 to t in a specific dataset
and Pt is the hidden activity levels of processes at time point t in the same dataset. In
our case, Pt is an n-dimensional column vector each dimension corresponding to a
process and Gi is an m-dimensional vector each dimension corresponding to a gene.
First, we compute the predicted mean and variance:
Pt|t 1 Pt 1|t 1
Vt|t 1 Vt 1|t 1 Q
where Q is the transition co-variance matrix. Next, we compute the following quantities:
et Gt WPt|t 1
St WVt|t 1W ' R
K t Vt|t 1W ' St 1
Lt log N (et , 0, St )
where W is our gene-process association weight matrix and R is the observation noise
matrix in emission model. Then, the estimates of mean and variance are:
Pt|t Pt|t 1 Kt et
Vt|t ( I KtW )Vt|t 1
Backward pass
First we compute:
Pt 1|t Pt|t
Vt 1|t Vt|t Q
J t Vt|tVt 1|t 1
Then, our expected activity levels of processes can be computed as:
Pt|T Pt|t J t ( Pt 1|T Pt 1|t )
By running a forward pass followed by a backward pass, we can infer the expected
activity levels for processes and other sufficient statistics. This inference algorithm is
performed for each dataset so that we can infer the hidden activity levels for each process
in all datasets.
For implementation, we used Kevin Murphy’s MATLAB BNT toolbox.
Learning:
When the weights are fully specified, CHPM resembles a factorial HMM [10] and
inference and learning can be performed by maximizing the expected complete log
likelihood. However, in our case the weights (and the edges corresponding to non zero
weights) are unknown. Since one of the goals of CHPM is to identify new process-gene
associations, we define a penalized complete log-likelihood score Score(o, h | W , )
where o, h represent all hidden and observed variables, respectively, and includes all
model parameters other than the association weights W.
D
n
m
n
Score(o, h : W , ) log( P(od , hd | W , )) 1 | wi , j |
d 1 j 1
m
n
i 1 j 1
2 ( wi , j 0)( Ei , j 1 (1 Ei , j ) 0 )
i 1 j 1
where hd and od are the unobserved activity levels for biological processes and the
observed expression levels for genes in dataset d, respectively.
Assuming the inferred expected activity levels of all biological processes are fixed, by
maximizing this score we can both determine the values for the parameters in the model
and the structure of the model. Let P j ,d ,t E ( Pj ,d ,t | G1:n ,d ,1:T , , W ) , i.e. the inferred
activity level of process j at time point t in dataset d, given all observed data and model
parameters. This can be inferred in E-step for each dataset independently. Then, the
penalized likelihood score can be rewritten as:
2
D n Td
P j ,d ,1 1
( P j ,d ,t P j ,d ,t 1 ) 2 1
2
Score(o, h : W , ) log(
log
)
log(
log 2 d )
d
2
2
2
2 d 2
2 d
d 1 j 1
d 1 j 1 t 2
D
n
n
D
m
Td
log(
(Gi ,d ,t wi , j P j ,d ,t ) 2
j 1
2 2 d
d 1 i 1 t 1
m
m n
1
log 2 d ) 1 | wi , j |
2
i 1 j 1
n
2 ( wi , j 0)( Ei , j 1 (1 Ei , j ) 0 ) C
i 1 j 1
where C is some constant not dependent on the parameters. Other notations are the same
as the main paper. By this score, we can estimate the variance terms 2 and the process
smoothness term 2 in closed form by zeroing the first derivative of the score defined
above. It’s easy to show that the updating rule is:
n
2
d
P
j , d ,1
j 1
n
n
2d
2
Td
( P
j , d ,t
P j ,d ,t 1 ) 2
j 1 t 2
n(Td 1)
After estimating 2 and 2 , we estimate the association weight matrix W and the
observation noise 2 d . Unlike other parameters, using MLE W and 2 d do not have a
closed form. Also, as we discussed in main paper, MLE of W will result in many nonzero or negative weight for each gene. Therefore, we use coordinate ascent to estimate W
and 2 d . In the first step, we fix W and estimate 2 d . In the second step, we fix 2 d and
estimate W. The second step has been discussed in detail in the main paper. Here, we
only give the updating rule for 2 d .
Assuming W is fixed, 2 d is updated by zeroing the first derivative of the score:
n
(
G
wi , j P j ,d ,t ) 2
i , d ,t
D m Td
score
1
j 1
2
0
log(
log
)
d
2
2 d
2 d d 1 i 1 t 1
2 2 d
Since we have two distinct noise terms a 2 d and b 2 d for each dataset representing the
noise for genes with and without incoming edges, respectively, these two terms can be
updated by considering only the corresponding set of genes:
a
2
d
b2d
Td
n
(Gi,d ,t wi, j P j ,d ,t )2
i{genes with edges} t 1
j 1
Td # genes _ with _ edges
Td
n
(Gi,d ,t wi, j P j ,d ,t )2
i{genes without edges} t 1
j 1
Td # genes _ without _ edges
Reference
[Jor02] M.I. Jordan. An introduction to probabilistic graphical models, 2002. In
preparation.
[Mur01] K.P. Murphy, The Bayes Net Toolbox for MATLAB (2001).
[Mur02] K.P. Murphy, Dynamic Bayesian Networks: Representation, Inference and
Learning, 2002. Ph.D. Thesis, UC Berkeley.