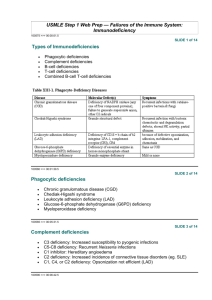
IMMUNOLOGY ا.م.د.هيفاء الحديثي Immunodeficiency When the
advertisement

هيفاء الحديثي.د.م.ا IMMUNOLOGY Immunodeficiency When the immune system fails to protect the host from disease-causing agents or from malignant cells, the result is ID. ID disorders secondary acquired Primary (congenital) Primary ID: 4 major immune components B-cell deficiencies T-cell deficiencies Phagocytic cell defect Complement deficiencies B-cell ID disorders 1. X-linked hypogammaglobulinemia (Bruton’s) low serum level of all classes of Ig (the level of Abs are absent or greatly reduced) due to failure in maturation of pre-B cells into B-Cells, also lymphoid organs are incompletely developed. The actual genetic background for this disease involves some types of mutation in the gene for an essential enzyme (Tyrosine Kinase) required by B cells to servive and mature. Clinically, recurrent pyogenic infections occur in infants at about 6months of age, when maternal Ab is no longer protective. Treatment with pooled gamma globulin reduces the number of infections. The transient hypogammaglobulinemia in infancy mainly resolve by its own by (16-30) months of age. 2. Selective immunoglobulin deficiencies: Decrease in the level of one Ig or more but with normal or increase level of other Igs. IgA class deficiency most prevalent (1/600) with normal quantities of B cells but unable to synthesize IgA. IgA deficiency result in sinus and lung infections. Patients with selective Igm deficiency or IgG subclasses deficiency are liable for recurrent sinopulmonary infections caused by pyogenic bacteria.. T-cell ID disorders 1. Thymic aplasia (digeorge’s syndrome) results when embryonic third & 4th pharyngeal pouches fail to develop, or associated with deletion in chromosome 22. Both thymus and the parathyroids fail to develop properly result is sever viral, fungal or protozoal infections. The most common presenting symptom is tetany due to hypocalcemia. Ab production is either normal or decreased. Thymus transplant may be of some benefit. 2. Chronic mucocutaneous candidiasis, these children who present with skin and mucous membrane candidal infection have a T-cell deficiency specifically for Candida albicans. Usually treated with antifungal drugs. Combined B-cell and T-cell deficiencies 1. Hyper-IgM syndrome: In this syndrome, sever, recurrent infections begin early in life. T-helper cells have a defect in the surface protein that interacts with CD40 on B-cell surface, this result in inability of Bcell to switch from production of IgM to other classes of Igs. 2. Sever combined Immunodeficiency disease (SCID) this either due to stem cell defect results in absence of T and B cells or the number of cells is normal but they do not function properly, some due to gene mutation or absence of certain enzymes as: - Adenosine deaminase and nucleosides phosphorylase deficiency. Lymphocytes develop but a metabolic product build up cells abnormally and selectively destroy them. - X-linked deficiency in IL receptors for T&B cells. - Wiskott-Aldrich Syndrom. - Ataxia-Telangiectasia. - Also deficient expressional MHC molecules may result in combined deficiencies as lack of gene that code for HLA, MHC I & MHC II (Bare-lymphocyte syndrome) Complement deficiency Deficiencies in components or functions, grouped as (early component, late component or alternative cascade deficiencies). These may result in: 1. Hereditary angioedema. Absence of C1-inhibitor C1 act on C4C4a vasoactive (C3a & C5a) capillary permeability and edema in several organs. 2. Recurrent infections Patient with C3 deficiency are particularly susceptible sepsis with pyogenic bacteria such as S. aurous. Those with reduced C6, C7 or C8 are prone to bacteremia with Neisseria meningitides or Neisseria gonorrhoeae. 3. Autoimmune diseases: Patient with C2 & C4 deficiencies have autoimmune diseases. C3 deficiency are associated with SLE. 4. Paroxysmal Nocturnal Hemoglubinurea: Episodes of brownish urine (hemoglobin urea) complement-mediated hemolysis caused by deficiency of decay-accelerating factor (DAF). Phagocytic cell defects 1. Quantitative defects a. Neutropenia increased destruction - Autoimmune phenomena following certain drugs - Hypersplenism from exaggerated destructive function. b. Asplenia - Congenital - Surgical - Malignancy - Sickle cell anemia decreased production By bone marrow suppression. Leukemia Inherited stem cells defect. 2. Qualitative defects Defect may involve any of the phagocytic activities (chemotaxis, ingestion or intracellular killing) as in: - Chronic granulomatous disease - G6PD deficiency . - Job’s syndrome ( characterized by increase IgE, sever atopic dermatitis& repeated pyogenic infections). - Lazy leukocyte syndrome - Chediak-Higashi syndrome ( abnormality in lysosomal granules) Secondary immune deficiencies 1. Malnutrition-Zinc deficiency -Vitamine deficiency 2. Systemic disorders-renal insufficiency -Extensive burns 3. Drug induced-alcohol -Opiates -Immune suppressive treatment during transplantation or cancer Postsurgery- Transient depression of immune function due to surgery itself or general anesthesia 4. Splenectomy 5. Thymetomy 6. Malignancy -NHL -B cell malignancy 7. Infectious diseases a. Bacterial infection (TB) IL10 & IL4 ↓ Th1 b. Parasitic infection (trypanosome Cruzi) ↓ CMI c. Measles & other viral infections Transient suppression of delayed hypersensitivity. d. Acquired immune deficiency syndrome (AIDS) Caused by human immunodeficiency virus (HIV) which interact with a large number of different cells in the body and escaping the host immune response against it. Transmit through sexual intercourse and contact with infected blood, and infected mothers can pass HIV to their infants. Phases (clinical stages) of HIV infection - Acute retroviral infection lasting from infection until onset of detectable virus-specific Ab. lasts 3-8 weeks (IMN like symptoms) - Asymptomatic phase: last for mouths to >15 years but viral replication continues and the immune response is active and virus escape. - Clinical progression to AIDS, the host immune response. Include early & late symptoms and followed by advanced infection (fullblown syndrome) HIV and immune system - Primary cellular target is CD4+, the initial interaction between HIV & CD4+ involves specific region (gp120) and CD4+, and this interact with a protein found on the surface of some immune cells as coreceptor accepting HIV (CCR5). The expression of CD4 and CCR5 is highest in memory T-cells, so their rapid loss leads to increasing ID. Reduced CD4 T cells may also result in an incomplete activation of CD8 T cells, resulting in a decreased ability to destroy virally infection cells. Also down regulation of HLA class I inactivation of CD8 T cells [During asymptomatic phase, cell mediated immunity is prominent and CD8+ & CD28+ cells release a soluble factor that inhibit the replication of HIV] - High mutation rates of HIV also allow virus to escape immune responses. - A stronger humeral response against HIV can be detected (neutralizing Ab against gp120 & gp41) but the virus does not expose the immune dominant region of gp120 invivo, preventing the effectivity of gp120 Ab. Diagnosis of HIV infection - Screening of anti-HIV Ab by ELISA - Confirmatory test by western blot to detect structural protein (P24) and envelope glycol-proteins *gp41 & pg120). - By PCR Treatment of AIDS Anti-retroviral drugs inhibit the growth and replication of HIV at various stages of its life cycle. Now highly active anti-retroviral therapies develop include combination of more than one inhibitor. (HAART). Attacking HIV with antiretroviral drugs