Cerebral vasospasm in SAH
advertisement

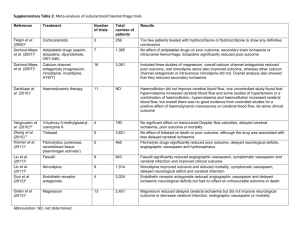
MEDICAL MANAGEMENT OF CEREBRAL VASOSPASM Mark Angle, M.D. 2001 i. What is “cerebral vasospasm”? The term is syndromic. It refers to the occurrence of regional brain hypoperfusion from 4 to 14 days after a sub-arachnoid hemorrhage. The condition frequently leads to delayed ischemic neurological deficits (DIND) and cortical infarction. (6, 7, 8) What are ‘delayed ischemic neurological deficits’? Again, the term is syndromic. It refers to the appearance of new neurological signs in the postictal period of SAH which cannot be explained by rebleeding, seizure, hydrocephalus, electrolyte disorder or concurrent medical problems. The symptoms may be focal (hemiparesis, aphasia) or just as often, global (lethargy, confusion, obtundation). ii. Is “cerebral vasospasm” vasospasm? Sometimes. The term was first used as a presumed explanation for the focal angiographic narrowing of proximal vessels within the Circle of Willis complex which usually accompanied delayed neurological deficits. Pathologically however, the lesion is not due to vasoconstriction but to an inflammatory arteritis with luminal compromise. That being said, there is likely some active vasoconstriction at the site of the inflammatory lesion, as well as some distally, within angiographically unassessable vessels. The latter explains why symptoms do not always correspond to the anatomic lesion nor respond to angioplasty. (21) What causes vasospasm? Notionally, oxyhemoglobin from the blood clot which invests the artery. Sub-arachnoid blood fills the CSF spaces to a greater or lesser extent, depriving vessels of CSF-borne nutrients and allowing the build-up of various pollutants. Products of erythrocyte breakdown contaminate the CSF circulation and may cause distant effects. Those local and circulating factors may be spasmogenic and/or pro-inflammatory. The identity of the factor, or more likely factors, linking oxyHb to vasospasm remains obscure despite 50 years of investigation. It is complicated by the identification of two phases of vasoconstriction, early and late, only the first of which is produced in animal models. In general terms, SAH causes an imbalance between vasodilating (NO cGMP), vasoconstricting (endothelin PKC) and eicosonoid (cAMP, 1P3) pathways. (1, 2, 5, 11, 24) Is the proximal lesion the only cause for regional ischemia? Other mechanisms have been proposed, though they amplify the consequences of large artery constriction. Evidence exists that the microcirculation is diffusely disordered post-SAH and responds abnormally to neuronal activation. Clot hemolysate in CSF has been shown to produce spreading cortical depression. Experimentally, this combination of factors produces cortical necrosis of a pattern indistinguishable from that seen clinically. (17) Can we predict the risk of “vasospasm”? Angiographic narrowing occurs in 50-70% of all patients post-SAH, with half of those becoming symptomatic. Since the incidence of clinically significant vasospasm is then almost 30%, predictive factors are interesting epidemiologically and perhaps pathophysiologically, less so clinically. The identified risk factors are: thickness of the clot in the cisterns, Sylvian fissures and lateral ventricles, GCS≤11, hydrocephalus, early surgical intervention, intra-operative hypotension and a prior history of smoking or hypertension. Only hemorrhages confined to the perimesencephalic spaces seem to be exempt from risk. (7, 21, 22, 23, 25) Can we reliably diagnose “vasospasm”? Almost by definition, regional hypoperfusion is best diagnosed by symptoms (as ‘retro’ as that may seem). Angiography remains the gold standard for identifying the vascular lesion, but it misses any distal component and fails to give regional blood flow. PET and MRI quantitation of perfusion remain research tools. CT-based perfusion techniques are promising, especially if they can be used to evaluate therapy. Surgically implanted microdialysis catheters are in use, but suffer from a too restricted region of interest. Jugular-bulb saturation monitoring suffers the opposite problem. Trans-cranial doppler (TCD) measures one derivative of flow – velocity – in proximal vessels. It’s accessible, repeatable and reproducible. Unfortunately, it’s not reliable as an independent marker in the absence of symptoms. TCD has no meaningful correlation with measures of regional perfusion. It’s sensitivity and specificity vary widely when compared to angiography, especially in the elderly. There is no evidence to support initiating treatment using TCD criteria alone. (10, 19) Why then do we do TCD’s? TCD helps confirm the etiology of delayed deficits i.e. a positive TCD in the presence of symptoms has good predictive power. A trend towards higher velocities as days pass is a reasonable indication to maintain close surveillance. Finally, in patients who have a poor baseline and in whom the neuro exam is insensitive, TCD provides reasonable indications to treat. Can “vasospasm” be prevented? Of all the factors which predict vasospasm and its consequences, only the timing and conduct of surgery is under our control. While supporting data is still equivocal, most centres have chosen early intervention as their practice. Intra-operative hypotension is now rarely used. The only clinical intervention promising to reduce the incidence of arterial narrowing is that of cisternal lavage with fibrinolytic agents. This requires the placement of a microcatheter either intra-operatively or by retrograde guidance from the lumbar interspaces. By actively clearing the clot, vasospasm seems to be prevented. A variety of drugs (NO donors, endothelin antagonists, K channel activators) have shown some attenuation of the vascular pathology in experimental models but none are in clinical use. (3, 4, 18, 27) If we can’t prevent “vasospasm”, can we prevent DINDs? The most effective therapy is probably eliminating the factors which worsen the consequences of the arterial narrowing. Prominent amongst these are volume contraction, hyponatremia, fever, anti-hypertensive drugs and the use of anti-fibrinolytics. For a variety of obvious reasons (inanition, surgical losses, dye-induced diuresis) patients are often hypovolemic at baseline. An aggravating factor, and one associated with vasospasm itself, is a profound natriuresis (urine output 2000 cc q8h, urinary Na+ 120-280 meq/L). Unchecked, this produces falls in cardiac output and BP, necessarily reducing flow across narrowed arterial segments and accentuating the hypoperfusion. Hyponatremia produces cerebral swelling and a fall in CPP. The root of this unique natriuretic state is a high level of brain natriuretic peptide (BNP) which can last for up to 14 days. In practice, euvolemia is best maintained with colloids and hypertonic saline. Mean blood pressure tends to rise during vasospasm, partly due to BNP, partly as a homeostatic attempt to maintain flow across the constricted artery. Attempts to blunt this response are counter-productive. Fever is very common after SAH and is also a marker for vasospasm. (An argument can been made that SAH/vasospasm is a systemic inflammatory response (SIRS)). Fever is an independent predictor of poor outcome (odds ratio 1.4 per day above 38.3C). While treating the fever is not known to alter outcome, it seems reasonable to try COX-2 inhibitors. (8, 9, 20) How about triple H therapy? It’s a cult. That doesn’t mean that it’s without foundation or that it’s ineffective , just that the claims and expectations in its name are exaggerated. It’s also catchy and easy to remember. The three H’s are hemodilution, hypervolemia and hypertension. The rationale is simple: if there is a fixed constriction, flow across it is a function of blood rheology and input pressure. Since collateral flow is subject to the same factors, that might be augmented in tandem. The independent effect of hemodilution is unclear, just as it is in stroke. In practice, the hct is maintained at 30, a figure associated with low effective viscosity. Hypervolemia is now difficult to justify. Early responses to large volume challenges probably reflected unrecognized hypovolemia at baseline. Euvolemia achieves 95% of the maximal cardiac output without the risks of cardio-respiratory events. Studies have shown no effect on cardiac output or cerebral blood flow at supra-normal filling pressures. Induced hypertension is an effective first step in treating neurological deficits. The usual protocol involves increasing MAP by 20-30 mmHg using phenylephrine or levophed. Failure to respond or recidivism would then lead to angiography/angioplasty. In practice, mild passive hemodilution and careful maintenance of circulating volume are applied to all patients “prophylactically’, and hypertension to those who are symptomatic. ADD: Using a CVP tracing, optimal filling pressures are achieved by volume expansion until the Y-descent is accentuated and the respiratory variation minimized. PA catheters are rarely helpful. ADD2: The AHA considers evidence for HHH to be class C as does the Cochrane Colloboration i.e. unproven. ADD3: Triple H therapy is associated with significant morbidity. (8, 13, 14, 15, 28) How does Nimodipine work? Nimodipine, alone amongst the Ca++ channel blockers, improves outcome (AHA Class I). The optimal dose and duration have not been established but 60mg po q4h is effective. The effect is a 33% relative risk reduction for the development of a neurological deficit, and a 20% relative risk reduction in cerebral infarction. To protect one patient, 8 patients have to be treated. Nimodipine does not alter the incidence or severity of angiographic narrowing. Its benefit then is either a vascular effect on distal vessels, a neuroprotective one (decreasing the ischemic threshold of neural tissue) or one related to its anti-platelet and anti-fibrinolytic actions. It is worth noting that the effect of Nimodipine is lessening in more recent studies, probably reflecting the evolution of neurological critical care. (8, 12) How effective is ‘best management’ of symptomatic vasospasm? Qureshi (John Hopkins) reports a 50% incidence of stroke 24 hrs after beginning therapy for symptomatic vasospasm, and a 44% poor outcome for the same group. Other studies corroborate these figures, despite the implementation of angioplasty protocols. (8, 25, 28, 29) Are any innovative therapies in-the-works? Many. Since almost everything works in the lab, we are likely to see NO donors, endothelin converting enzyme inhibitors, K-ATP channel openers, PKC inhibitors, estradiol, milrinone and gene therapies. Milrinone for “vasospasm”? Isn’t it obvious? A PDEIII inhibitor, milrinone affects both cAMP and cGMP pathways (through cross-talk) to accomplish vasodilation. In our lab, milrinone bypasses the endothelium to inhibit pathological constriction in a number of micro-environments. It is an important immunomodulator, inhibiting TNF and IL-6, as well as influencing wbc and platelet function. Does it work? Milrinone was first reported to inhibit the development of vasospasm in dogs in 1996. A recent study reports its use in 7 patients in the cath lab, continuing through their ICU stay, all with encouraging outcomes. At the MNH, we have used milrinone trials in symptomatic vasospasm since April 1999. Twenty-eight consecutive patients have been treated without adverse effects. In this group, none deteriorated, most (26/28) improved, only 2 required hypertensive therapy, one died of an unrelated hepatic condition, two CT hypodensities developed and only two patients were dependent at 6 months. (16) Any idea how it works? In the 8 patients who received milrinone in the angio suite, 6 showed obvious changes in large vessel caliber but all 8 showed enhanced flow. This suggests a concurrent decrease in downstream i.e. small vessel resistance. In the ICU, no reliable change in TCD velocities was detected. Only 2 patients had cardiac output measurements and neither changed dramatically. What is the future for Milrinone? Milrinone, like the other agents, should be subjected to a PCRT to establish efficacy and an effect on outcome. Its high safety index also makes testing as a prophylactic agent desirable. Recommendations? 1. No intra-operative hypotension (intentional or otherwise) 2. CVP – guided normovolemia 3. Aggressive temperature control 4. Maintenance of normal serum Na+ 5. No antihypertensives 6. Symptom – guided interventions i. milrinone trial ii. hypertensive trial iii. angioplasty with papaverine trial 7. Continue searching for the truth while getting through the day with workable beliefs… 1. Dietrich H.H. et al: Molecular Keys to the Problems of Cerebral Vasospasm. Neurosurgery 46: 517-530, 2000 2. Juvela S.: Plasma endothelin concentrations after aneurysmal subarachnoid hemorrhage. J. Neurosurgery 92: 390-400, 2000 3. Kwan A.-L. et al: Continuous Intravenous Infusion of CGS 26303, an Endothelin-converting Enzyme Inhibitor, Prevents and Reverses Cerebral Vasospasm after Experimentail Subarachnoid Hemorrhage. Neurosurgery 49: 422-429, 2001 4. Kiris T. et al: Reversal of Cerebral Vasospasm by the Nitric Oxide Donor SNAP in an Experimental Model of Subarachnoid Haemorrhage. Acta Neurochir (Wien) 141: 1323-1329, 1999 5. Khaldi A. et al: Measurement of Nitric Oxide and Brain Tissue Oxygen Tension in Patients after Severe Subarachnoid Hemorrhage. Neurosurgery 49: 33-40, 2001 6. Kassell N.F. et al: The International Cooperative Study on the Timing of Aneurysm Surgery. J. Neurosurgery 73: 37-47, 1990 7. Longstreth Jr, W.T. et al: Clinical course of spontaneous subarachnoid hemorrhage. Neurology 43: 712-718, 1993 8. Mayberg M.R. et al: Guidelines for the management of Aneurysmal Subarachnoid Hemorrhage. Stroke 25: 2315-2328, 1994 9. Roos Y.: Antifibrinolytic treatment in subarachnoid hemorrhage. Neurology 54: 77-82, 2000 10. Torbey M.T.: Effect of Age on Cerebral Blood Flow Velocity and Incidence of Vasospasm After Aneurysmal Subarachnoid Hemorrhage. Stroke 32: 2005-2011, 2001 11. Osuka K. et al: Interleukin-6 and Development of Vasospasm after Subarachnoid Haemorrhage. Acta Neurochir (Wien) 140: 943-951, 1998 12. Feigin V.L. et al: Calcium antagonists in patients with aneurysmal subarachnoid hemorrhage. Neurology 50: 876-883, 1998 13. Pritz M.B.: Treatment of Cerebral Vasospasm due to Aneurysmal Subarachnoid Hemorrhage: Past, Present, and Future of Hyperdynamic Therapy. Neurosurgery Quarterly 7: 273-285, 1997 14. Lennihan L. et al: Effect of Hypervolemic Therapy on Cerebral Blood Flow After Subarachnoid Hemorrhage. Stroke 31: 383-391, 2000 15. Egge A. et al: Prophylactic Hyperdynamic Postoperative Fluid Therapy after Aneurysmal Subarachnoid Hemorrhage: A Clinical, Prospective, Randomized, Controlled Study. Neurosurgery 49: 593-606, 2001 16. Arakawa Y. et al: Milrinone for the Treatment of Cerebral Vasospasm after Subarachnoid Hemorrhage: Report of Seven Cases. Neurosurgery 48: 723-730, 2001 17. Dreier J.P. et al: Products of hemolysis in the subarachnoid space inducing spreading ischemia in the cortex and focal necrosis in rats: a model for delayed ischemic neurological deficits after subarachnoid hemorrhage? J. Neurosurg 93: 658-666, 2000 18. Kodama N. et al: Cisternal Irrigation Therapy with Urokinase and Ascorbic Acid for Prevention of Vasospasm after Aneurysmal Subarachnoid Hemorrhage. Surg Neurol 53: 110-118, 2000 19. Nilsson O.G. et al: Bedside Detection of Brain Ischemia Using Intracerebral Microdialysis: Subarachnoid Hemorrhage and Delayed Ischemic Deterioration. Neurosurgery 45: 1176-1185, 1999 20. Sviri G.E.: Brain Natriuretic Peptide and Cerebral Vasospasm in Subarachnoid Hemorrhage. Stroke 31: 118-122, 2000 21. Mayberg, Marc. Neurosurgery Clinics of North America. Vol. 1(2). Philadelphia: W.B. Saunders Company, 1990 22. Dorsch, N.W.: A Review of Cerebral Vasospasm in Aneurysmal Sub-Arachnoid Hemorrhage Part I: Incidence and Effects. 23. Chang, H.S. et al: Adverse effects of limited hypotensive anesthesia on the outcome of patients with subarachnoid hemorrhage. J. Neurosurg. 92: 971-975, 2000 24. Laher I. et al: Protein Kinase C and Cerebral Vasospasm. Journal of Cerebral Blood Flow and Metabolism 21: 887-906, 2001 25. Claassen J. et al: Effect of Cisternal and Ventricular Blood on Risk of Delayed Cerebral Ischemia After Subarachnoid Hemorrhage. Stroke 32: 2012-2020, 2001 26. Brilstra E.H. et al: Rebleeding, secondary ischemia, and timing of operation in patients with subarachnoid hemorrhage. Neurology 55: 1656-1660, 2000 27. Kwan A.-L. et al: Delayed Administration of the K+ Channel Activator Cromakalim Attenuates Cerebral Vasospasm after Experimental Subarachnoid Hemorrhage. Acta Neurochir (Wien) 142: 193-197, 2000 28. Qureshi A.I. et al: Early predictors of outcome in patients receiving hypervolemic and hypertensive therapy for symptomatic vasospasm after subarachnoid hemorrhage. Crit Care Med 28: 824-829, 2000 29. Clarke Haley E. et al: A randomized, double-blind, vehicle-controlled trial of tirilazad mesylate in patients with aneurysmal subarachnoid hemorrhage: a cooperative study in North America. J. Neurosurg 86: 467-474, 1997