Lecture Slides
advertisement

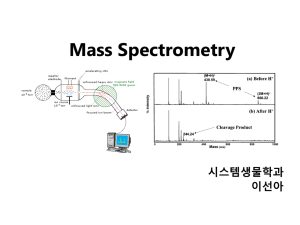
Mass spectrometry-based proteomics Jeff Johnson Feb 19, 2014 MS Proteomics in a Nutshell • Ionization – Delivering macromolecules to the MS • Ion Manipulation – Trapping and ejecting analytes of interest • Fragmentation – Breaking apart for more information • Mass analysis and detection – Measuring masses and quantifying intensities MS Proteomics in a Nutshell • Ionization – Delivering macromolecules to the MS • Ion Manipulation – Trapping and ejecting analytes of interest • Fragmentation – Breaking apart for more information • Mass analysis and detection – Measuring masses and quantifying intensities Macromolecular Ionization for MS • Analyte must be in the gas phase for mass analysis • Analyte must be charged in order to be manipulated by electric and magnetic fields – All mass analyzers measure mass-to-charge ratios (m/z) • Two predominant approaches (shared the Nobel prize in 2002) – Matrix assisted laser desorption ionization – Electrospray ionization MALDI Ionization • Sample is spotted in a matrix that readily absorbs UV/IR light and is vaporized by a laser – Common matrix: 2,5dihydroxybenzoic acid (DHB) • Advantages – Fast and easy – Spots can be reanalyzed later – Most analytes get one +ive charge makes it easy to deconvolute • Disadvantages – Harsh. Often breaks analytes apart (e.g., breaks phosphorylation) – Not easily combined with online HPLC separations Electrospray Ionization • Sample is dissolved in liquid and pushed through a charged needle and sprayed into an evaporation chamber – Droplets pulled into the MS source by electric potential between the needle and the MS – Heated ion transfer tube evaporates water molecules in droplets leaving +ively charged analytes in the gas phase • Advantages – Compatible with online HPLC separations – “Soft” ionization maintains label and noncovalent interactions • Disadvantages – Analytes can have different numbers of charges, can be difficult to deconvolute without high mass accuracy – Different samples going through the same electrospray tip causes carryover problems • Especially bad with online HPLCs Ionization is Nearly Impossible to Predict • Different molecules ionize with different efficiencies and are very difficult to predict • MS intensity ratios between different molecules do not reflect ratios in the sample from which they were derived • Most quantification by MS is relative 2x A A X B B Ionization is Nearly Impossible to Predict Sample 1 Sample 2 A A Sample 1 Sample 2 A A • Different molecules ionize with different efficiencies and are very difficult to predict • MS intensity ratios between different molecules do not reflect ratios in the sample from which they were derived • Most quantification by MS is relative * Assumption: MS run 1 = MS run 2 MS Proteomics in a Nutshell • Ionization – Delivering macromolecules to the MS • Ion Manipulation – Trapping and ejecting analytes of interest • Fragmentation – Breaking apart for more information • Mass analysis and detection – Measuring masses and quantifying intensities Ion Manipulation • We need a way to select only ions of interest – Most detectors are just electron multipliers that don’t measure mass but just detect a thing hitting the multiplier – We can manipulate ions to deliver defined mass ranges to the detector to get a mass spectrum • Two common tools: – Ion traps – Quadrupoles – Both use electric and magnetic fields to select ions of a particular m/z range Ion Trap • Ions are trapped by 3D electric field by DC and AC applied to the electrodes • An ion trap can accumulate ions as they come in from the source and store them • Low resolution: +/- 1 Da Quadrupole • Can be thought as a mass filter • DC and AC fields applied that stabilize a trajectory for ions in a desired mass range, undesired ions are ejected • Quadrupole operate with a continuous flow of ions • Low resolution (+/- 1 Da) MS Proteomics in a Nutshell • Ionization – Delivering macromolecules to the MS • Ion Manipulation – Trapping and ejecting analytes of interest • Fragmentation – Breaking apart for more information • Mass analysis and detection – Measuring masses and quantifying intensities Fragmentation • Usually measuring the mass of an analyte is not enough to conclusively identify it • By fragmenting an analyte and measuring the masses of the fragments we can obtain further information to identify the analyte • There are many types of fragmentation but collision-induced dissociation (CID) is the most common – Fastest and most generally successful for the widest variety of proteins and peptides Collision-Induced Dissociation • Give ions kinetic energy and collide with gas molecules (He) • Collisions build up potential energy until a fragmentation event can occur • Ideally potential energy is strong enough to break a single peptide bond but not strong enough to fragment further • Can be done in an ion trap or a quadrupole Collision Induced Dissociation A E P T I R H2 O Fragment (somewhat) randomly along the peptide backbone Intensity B-type Ions A E P A E P A E A 201.1 T 298.1 399.2 72.0 M/z Y-type Ions R I T Intensity H2 O M/z P E A B-type, A-type, Y-type Ions R I T Intensity H2 O M/z P E A MS Proteomics in a Nutshell • Ionization – Delivering macromolecules to the MS • Ion Manipulation – Trapping and ejecting analytes of interest • Fragmentation – Breaking apart for more information • Mass analysis and detection – Measuring masses and quantifying intensities Mass Analysis and Detection • All mass analyzers achieve the same thing: physical separation based on mass:charge • Magnetic sector is the simplest and one of the earliest types Magnetic Sector MS FT-ICR MS • FT-ICR = Fourier transform – ion cyclotron resonance • Ion injected in line with a strong magnetic field that induces a cyclical motion • Radius of the cyclotron motion is proportional to m/z Time-of-flight MS Medium / High Resolution Quadrupole and Ion Trap MS Electron multiplier • You can use a quadrupoles or ion traps to “scan out” ions across an entire mass range to a detector by gradually ramping voltages • Low resolution but electron multipliers make these very sensitive Orbitrap MS • Characteristic frequencies: – Frequency of rotation ωφ – Frequency of radial oscillations ωr – Frequency of axial oscillations ωz r z z 2 Rm 1 2 R 2 r z Rm 2 R k z m/ z φ U (r , z ) k 2 z r 2 / 2 Rm2 ln( r / Rm ) 2 Power of Fourier Transforms • FTs convert from time domain to freq domain • Instead of a single measurement the m/z is measured over a period of time and the FT essentially averages all those measurements • Resulting data is very high resolution Chromatography to Simplify Complexity Complex Sample MS • Complexity hurts sensitivity • A constant, defined number of ions can be analyzed in each MS scan • Sensitivity is constant (around 1 fmol) • A scan with fewer ions is more sensitive than a scan with many Chromatography to Simplify Complexity HPLC Complex Sample MS C18 RP column ACN gradient A C B A B D C D Chromatography to Simplify Complexity Very Complex Sample Offline HPLC (e.g., SCX) SCX Fractions Injected individually Online HPLC (RP) SCX Fractions MS Acquiring MS Data • Data acquisition depends on experimental goals – Data-dependent acquisition • MS attempts to acquire data to allow you to identify a maximum number of unknowns • Commonly used for analyses where you don’t know what you’re looking for – Targeted acquisition • MS only acquires data for what you tell it to acquire • Much more sensitive than data-dependent, but also more limited in scope Data-Dependent Acquisition Data-Dependent Acquisition 1 3 2 High resolution survey scan (<5 ppm mass accuracy) Data-Dependent Acquisition Low resolution MS/MS scan 1 Data-Dependent Acquisition Low resolution MS/MS scan 2 Data-Dependent Acquisition Low resolution MS/MS scan 3 Peptide Identification AA sequence DB (Species UniProt) Peptide Identification AA sequence DB (Species UniProt) 1 2 3 AA DBs restricted by parent ion mass measured in survey scan Peptide Identification AA sequence DB (Species UniProt) MS/MS 1 1 MS/MS 2 2 MS/MS 3 3 AA DBs restricted by parent ion mass measured in survey scan # of Matches Probabilistic Matching (X!Tandem) Second Best Best Hit by-Score= Sum of intensities of peaks matching B-type or Y-type ions Hyper Score HyperScore= ( by-Score) × N ! × N ! y b Model spectrum comparisons Pattern Matching (Sequest) Sequest XCorr Correlation Score Cross Correlation (direct comparison) Auto Correlation (background) Offset (AMU) XCorr = CrossCorr avg AutoCorr offset=-75 to 75 ( ) Targeted Acquisition with a QQQ SRM Assay Peptide Fragment Q1 Q3 RT Rel Int YGFIEGHVVIPR y8_1 693.88 906.52 22.9 100 YGFIEGHVVIPR y7_1 693.88 777.47 22.9 91 YGFIEGHVVIPR b3_1 693.88 368.16 22.9 77 YGFIEGHVVIPR y4_1 693.88 484.33 22.9 53 YGFIEGHVVIPR y3_1 693.88 385.26 22.9 42 A priori knowledge required: SRM assay development for a list of proteins/peptides of interest Information derived from label-free unbiased proteomic analysis “Sensitivity” • Sensitivity of a MS is well defined, but the ability to identify something is a very different concept – Ability to detect depends on: • Sample complexity • MS sensitivity • MS speed – A faster MS can collect go deeper in each survey scan – Think “top 10” vs. “top 50” • MS mass accuracy – Better mass accuracy improves the ability to identify peptides but sacrifices speed and MS sensitivity – Especially important for variable modifications – The “best” method is very dependent on the experimental goals Database Searching Ion trap +/- 1 Da Orbitrap +/- 0.002 Da Database “search space” Database Searching Ion trap +/- 1 Da Database “search space” +S/T/Y phosphorylation Orbitrap +/- 0.002 Da Database Searching Ion trap +/- 1 Da Database “search space” +S/T/Y phosphorylation Orbitrap +/- 0.002 Da Protein Quantification • A mass spectrometer is an inherently quantitative device but the ionization source is not – Different peptides/proteins are ionized with drastically different efficiencies – Absolute abundances in a mass spectrometer are not precisely indicative of abundance in a sample • Solution: stable isotope labeling – Compare samples that have been labeled with stable isotopes (13C, 15N, 2H) – ‘Heavy’ isotopes behave chemically identically to their ‘light’ counterparts but are separated in the MS Isotope Coded Affinity Tag (ICAT) Stable Isotope Labeling of Amino Acids in Culture (SILAC) • Grow cells in media supplemented with stable isotope-labeled amino acids • Combine samples at the level of cells and process as one sample • Minimize variability between samples for lysis and digestion • Different samples separated by mass in the MS Absolute Quantification (AQUA) Absolute Quantification (AQUA)