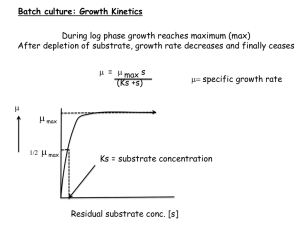
the Presentation - Pacific Northwest Biosciences Society
advertisement

In vitro and in vivo metabolism of repaglinide: Modeling clinicallyrelevant drug-drug interactions Joanna Barbara, Ph.D. Director of Analytical Services, XenoTech LLC. Pacific Northwest Biosciences Winter Seminar March 3, 2014 XenoTech’s integrated service capabilities XT Consulting Department Expert data review and study consultation Drug Metabolism Enzyme Induction Metabolic stability and species comparison In vitro studies in cultured hepatocytes Metabolite characterization/ID (human and animal) Reaction phenotyping (CYP & UGT) Ex vivo studies in animals Bioanalytical Customized services Toxicity & mechanistic studies Non-GLP Bioanalysis Enzyme Inhibition GLP and non-GLP in vitro study support Transporters XenoTech and Sekisui Evaluate potential for direct, and metabolismdependent inhibition(MDI or TDI) In vitro studies in mono-layer cell lines for uptake Mechanistic studies (direct or MDI) Bi-directional assay for efflux transporters Non-CYP enzymes (e.g., UGT, MAO, AO) Membrane-based vesicles and ATPase assays Sekisui Medical (Conducted in Japan) RI synthesis (radiolabeling), preclinical in vivo PK studies, QWBA, plasma protein binding, humanized chimeric mice (PXB), biomarker analysis, pharmacological receptor assays Products 2 Overview • Enzymatic biotransformation and drug-drug interactions • Introduction to repaglinide and project background – Repaglinide as a probe substrate • Investigating mechanism of drug-drug interactions – In vitro metabolism • Evaluating rat as a preclinical model – In vivo metabolism • Conclusions 3 Enzymatic biotransformation of drugs • Cytochrome P450 (CYP) enzymes are responsible for biotransformation of ~70% hepatically-cleared drugs Esterase CY P UG T UGT e ste ra se FM O CYP NA T M AO Hepatic clearance route by enzyme type Data adapted from: Cassarett and Doull’s Toxicology (2001) C. Klaassen (Ed), New York, NY: McGraw-Hill 4 Drug-drug interactions (DDI) • Altered enzymatic biotransformation can lead to clinically-relevant drug-drug interactions between coadministered drugs, a key safety consideration • In preclinical drug development, DDI risk is assessed by evaluating – Major clearance routes (e.g., mass balance, CYP phenotyping) – Enzyme inhibition potential – Enzyme induction potential – Transporter involvement and inhibition potential 5 Cytochrome P450 inhibition • CYP inhibition has potential to result in – Black box label warnings – Withdrawal from market O O O N F N N H N OH Terfenadine: withdrawn 1997 (victim drug) Mibefradil: withdrawn 1998 (perpetrator drug) Co-administration with CYP3A4 inhibitors (e.g., ketoconazole) reduced clearance of the drug and resulted in cardiotoxicity caused by terfenadine accumulation Mibefradil inhibits CYP3A4 and can cause elevated levels of coadministered drugs cleared by these enzymes. Life-threatening interactions can occur with bblockers and other antihypertensives 6 Repaglinide uses • Repaglinide is an insulin secretagogue used to normalize postprandial hyperglycemia in patients with type 2 diabetes OH OH CH3 O H 3C O NH CH3 CH3 O H3C O O NH O CH3 O N NH Repaglinide OH Dicarboxylic acid (M2) • Major human metabolite in vivo is the dicarboxylic acid (van Heiningen et. al., 1999) • Other oxidative metabolites and glucuronide conjugate van Heiningen et al. Eur, J. Clin. Pharmacol. Exp. Ther. 1999; 55(7): 521-525. 7 Repaglinide metabolism • Major biotransformation routes described (Bidstrup et al., 2003) CYP2C8 metabolism M0-OH M4 CYP2C8 probe CYP3A4 metabolism M1 M2 M5 Bidstrup et al. Br. J. Clin. Pharmacol. 2003; 56: 305-314. 8 Repaglinide M4 formation and antibody inhibition Roles for CYP2C8 CYP3A4 Bidstrup et al. Br. J. Clin. Pharmacol. 2003; 56: 305-314. 9 Repaglinide metabolized by CYP3A4/2C8 and UGT1A1 • Repaglinide therefore has potential for DDIs with other drugs cleared hepatically by CYP3A4 and 2C8 and UGT1A1 • According to the University of Washington Drug Interaction Database, repaglinide is known for interactions with 10 drugs – Flucloxacillin and rifampin cause increased CL – Gemfibrozil, clarithromycin, cyclosporine, deferasirox, telithromycin, itraconazole, trimethoprim cause >40% increase in AUC 10 Gemfibrozil and repaglinide • Type 2 diabetics have 2-4-fold increased risk of macrovascular disease • Gemfibrozil is used to reduce triglycerides (TG) in patients with certain dyslipidemias – Almost 30% TG reduction in diabetics compared to placebo group Vinik and Colwell Diabetes Care 1993; 16(1): 37-44. • In patients concommitant administration has resulted in up to 8-fold plasma increase in repaglinide Backmann et al. Drug Metab. Dispos. 2009; 37(12): 2359-66. – Reports of severe, prolonged hypoglycemia 11 Gemfibrozil dosing and pharmacokinetics • Gemfibrozil usually dosed at 600 mg twice a day or less commonly 900 mg once daily • PK parameters after a single oral dose Parameter 600 mg dose 900 mg dose Cmax (µg mL-1) 28.8 ± 4.1 40.8 ± 12.6 tmax (h) 1.8 ± 0.8 1.8 ± 0.8 AUC0-8 (µg h mL-1) 80.3 ± 10.3 132.1 ± 35.3 CL (L-1) 7.1 ± 0.9 6.6 ± 1.6 Vd (L-1) 11.6 ± 2.1 12.5 ± 3.4 t1/2 (h) 1.1 ± 0.2 1.3 ± 0.1 Rouini et al. Int. J. Pharmacol. 2006; 2: 75-78. 12 Gemfibrozil metabolism • Metabolized in liver to 4 major metabolites but the glucuronide metabolite is a potent CYP2C8 inhibitor Baer et al. Chem. Res. Toxicol. 2009; 22(7): 1298-1309. 13 In vitro experiments with repaglinide • Initially worked to establish a simple CYP2C8 assay in vitro to complement the in vivo application of repaglinide • Noted discrepancies using reference material potential issues with some of the analytical work described in the literature • Subsequently needed to re-establish the specificity of the CYP2C8/CYP3A4 metabolism 14 Repaglinide in human liver microsomes (HLM) • High-resolution LC UV chromatogram (254 nm) HLM-30 min RD117007_MSE_05Apr12_008 Sb (3,40.00 ); Sm (SG, 40x1) 4: Diode Array 50 mM Repaglinide 254 Range: 1.102e-2 0.5 mg/mL HLM 30 minutes; 37°C; pH 7.4 NADPH-generating system 8.72 Repaglinide 9.0e-3 8.0e-3 7.0e-3 6.0e-3 AU 5.0e-3 4.0e-3 3.0e-3 2.0e-3 1.0e-3 Probe metabolite Hydroxyrepaglinide (M4) Major in vivo metabolite Repaglinide dicarboxylic acid metabolite (M2) Repaglinide desaturation metabolites 17.62 17.24 0.0 High abundance Low abundance -1.0e-3 Time 6.00 8.00 10.00 12.00 14.00 16.00 Unlabeled peaks are not related to repaglinide 15 Repaglinide human liver microsome metabolite profile Component tr (min) m/z value Mass error (ppm) Mass shift Proposed biotransformation M0-OH 5.48 469.2693 -1.9 +15.9940 Hydroxylation C1 5.94 451.2590 -0.2 -2.0156 Dehydrogenation C2 6.83 441.2369 4.7 -12.0384 O-deethylation + hydroxylation M4 7.50 469.2700 -0.4 +15.9947 Hydroxylation M1 7.95 385.2112 -3.9 -68.0641 N,N-didealkylation M5 8.26 425.2436 -4.5 -28.0317 O-deethylation Repaglinide 8.98 453.2738 -3.3 -0.0015 None C3 10.09 471.2848 -0.9 +18.0095 Hydroxylation+ reduction M2 10.30 485.2642 -5.1 +31.9889 C4 16.09 451.2583 -3.1 -2.0170 N-dealkylation + oxidation to the carboxylic acid Dehydrogenation C5 17.23 451.2591 -1.3 -2.0162 Dehydrogenation C6 17.52 451.2599 0.4 -2.0154 Dehydrogenation 16 Recombinant CYP panel for repaglinide substrate loss • Incubating drug with individual enzymes can help narrow down enzymes involved in metabolism • Complicated by involvement of enzymes that would not be involved in a more complete test system Percentage of maximum remaining(%) 100 80 60 40 CYP3A4? Substrate loss 10 mM repaglinide 10 pmol/inc rCYP 20 minutes 35°C; pH 7.4 20 0 17 Inhibition experiments for CYP reaction phenotyping • Simple test system to minimize variables – HLM for cytochrome P450-mediated M0-OH, M1, M2, M4 and M5 • Use known chemicals (or antibodies) to inhibit specific enzymes – Mibefradil for CYP3A4 (metabolism-dependent) – Gemfibrozil glucuronide for CYP2C8 (metabolismdependent) • Assess the effect of the presence/absence of the inhibitor on formation of the metabolite of interest 18 Selecting appropriate conditions for inhibition experiments • Initial-rate conditions desirable 19 Metabolism-dependent CYP2C8 and 3A4 inhibition 2C8 3A4 Less clear 110 No inhibitor 100 Percentage of control (%) 90 Mibefradil 80 Gemfibrozil glucuronide 70 60 50 40 30 20 10 0 C1 (M0-OH) M0-OH C4 (M4) M4 C5M1 (M1) C7M5 (M5) Repaglinide metabolite C11 (M2) M2 20 Correlation data for major metabolites with HLM donor panel 21 Exploring the interaction further • Nonclinical species have very limited use in modeling human DDIs • One major challenge is species differences in protein expression and function (e.g., enzyme specificity) • Rodent studies occur early on for most drugs • Rat is not a good model for drugs cleared by CYP3A4 – Ortholog CYP3A1 has limited similarity and little overlap in function • The rat ortholog for CYP2C8 is CYP2C22 which has demonstrated some very similar properties • Could this DDI be modeled in the rat? 22 In vivo experiments in the rat (Xenometrics/XenoTech) 23 Repaglinide PK data in rat (n = 3 per group) Repaglinide plasma concentration (ng/mL) • AUC increase in group 1 animals Group 1: Gemfibrozil + repaglinide Group 2: Repaglinide only • Gemfibrozil concentrations 16 – 125 µg mL-1 24 Repaglinide PK data in rat (n = 3 per group) • Clear evidence of drug-drug interaction between gemfibrozil and repaglinide in Group 1 animals Parameter Group 1 Group 2 Fold-change Cmax (ng mL-1) 284.1 ± 85.2 66.2 ± 6.9 4.3-fold increase tmax (h) 1.7 ± 0.6 1.2 ± 0.7 1.4-fold increase AUC0-12 (ng h mL-1) 853.9 ± 344.7 242.8 ± 32.5 3.5-fold increase AUC0-∞ (ng h mL-1) 837.5 ± 337.0 282.1 ± 43.5 3.0-fold increase CLobs (L h-1 kg-1) 1299.3 ± 522.9 3622.0 ± 589.6 2.8-fold decrease Vdobs (L kg-1) 5158.6 ± 3397.4 13154.8 ± 2483.3 2.6-fold decrease t1/2 (h) 2.6 ± 0.8 2.6 ± 0.6 None 25 Repaglinide rat plasma (AUC pool) metabolite profile Component tr (min) m/z Proposed biotransformation Group 1 Group 2 RP1 3.10 441 O-Deethylation + hydroxylation + + M0-OH 5.58 469 Hydroxylation + ND RP2 5.78 469 Oxidation + ND Repaglinide glucuronide 7.17 629 Glucuronidation + + M4 7.42 469 Hydroxylation ND ND M1 7.81 385 N,N-Didealkylation ND ND RP3 7.87 441 O-Deethylation + hydroxylation + ND M5 8.30 425 O-deethylation + + RP4 8.59 451 Dehydrogenation + ND RP5 8.59 469 Repaglinide 8.89 453 Oxidation None + + ND + C3 9.74 471 Hydroxylation+ reduction + ND M2 10.12 485 N-dealkylation + oxidation to the carboxylic acid + ND 26 Relative abundance of major human metabolites Percentage of observed maximum (%) • Very low abundance metabolites in plasma • Limited plasma sample volume Plasma 100% 90% 80% 70% 60% 50% 40% 30% 20% 10% 0% Group 1 Group 2 27 Bile metabolite profiles (0-12 h pools) • Repaglinide predominantly excreted in bile in humans – 90% excreted in feces; 8 % excreted in urine van Heiningen et al. Eur, J. Clin. Pharmacol. Exp. Ther. 1999; 55(7): 521-525. • Rat bile profiles contained 49 metabolites across the two groups – Oxidative metabolism – Glucuronidation – Sulfonation • Initial focus has to be on metabolites of interest 28 Exploring the CYP inhibition in bile • Relative abundance of CYP2C8 (in human) metabolites decreased (~65%) with gemfibrozil dosing Percentage of observed maximum (%) 100% 90% 80% 70% 60% 50% Group 1 40% Group 2 30% 20% 10% 0% M0-OH M4 29 Relative abundance of major human metabolites Percentage of maximum observed (%) • All of them decreased with gemfibrozil dosing Bile 100% 90% 80% 70% 60% 50% 40% 30% 20% 10% 0% Group 1 Group 2 • Not characteristic of a specific CYP inhibition interaction 30 Urine metabolite profiles (0-12 h pools) • Huge differences between the treatment groups – Without gemfibrozil treatment, only 7 metabolites – With gemfibrozil treatment, 27 metabolites Component tr (min) m/z Proposed biotransformation Group 1 Group 2 Hydroxylation + ND Glucuronidation + ND 469 Hydroxylation + ND 7.83 385 N,N-Didealkylation + + M5 8.30 425 O-Deethylation + ND Repaglinide 8.89 453 None + + M2 10.12 485 N-dealkylation + oxidation to the carboxylic acid + + M0-OH 5.58 469 Repaglinide glucuronide 7.17 629 M4 7.42 M1 31 Metabolite abundance in the urine Percentage of maximum observed (%) • Even the metabolites detected in Group 2 urine are present at relatively low abundance Urine 100% 90% 80% 70% 60% 50% 40% 30% 20% 10% 0% Group 1 Group 2 32 Biliary vs urinary excretion • Gemfibrozil increases urine and decreases bile excretion 100 90 80 70 60 50 40 30 20 10 0 • Why? Bile Urine Percentage of total material (%) Percentage of total material (%) Group 1 Group 2 100 90 80 70 60 50 40 30 20 10 0 Bile Urine 33 Systemic effects of gemfibrozil • Metabolism-dependent CYP2C8 inhibitor – Does not seem to account for all the metabolic profile changes – As yet, do not have evidence of CYP2C22 inhibition • UGT1A1 inhibitor Gan et al. Br. J. Pharmacol. 2010; 70(6): 870-80. – Repaglinide glucuronidation occurs at least in part through 1A1 mediation – Would not explain other effects • OATP1B1 (SLCO1B1) hepatic uptake transporter inhibitor Nakagomi-Hagihara et al. Xenobiotica 2007; 37(5): 474-486. – Would severely reduce abundance of all metabolites in bile – May also account for increased urinary excretion in Group 1 34 Human and rat OATPs • Human OATP1B1 inhibition has been described as a confounding factor in the repaglinide/gemfibrozil DDI Kudo et al. Drug Metab. Dispos. 2012; 41(2): 362-371. • OATP1B family comprises OATP1B1 and 1B3 • Only rodent ortholog for OATP1Bs is Oatp1b2 – Functions similarly to both – Mice deficient in Oatp1b2 have shown some utility as models for OATP1B studies • Repaglinide PK has been shown to correlate with OATP1B1 polymorphism Niemi et al. Clin. Pharmacol. Ther. 2005; 77(6): 468-478. Kallioski et al. Br. J. Clin. Pharmacol. 2008; 66(6): 818-825. 35 Back to the PK data • The observed clearance, volume of distribution and t1/2 data do support the transporter hypothesis Parameter Group 1 Group 2 Fold-change Cmax (ng mL-1) 284.1 ± 85.2 66.2 ± 6.9 4.3-fold increase tmax (h) 1.7 ± 0.6 1.2 ± 0.7 1.4-fold increase AUC0-12 (ng h mL-1) 853.9 ± 344.7 242.8 ± 32.5 3.5-fold increase AUC0-∞ (ng h mL-1) 837.5 ± 337.0 282.1 ± 43.5 3.0-fold increase CLobs (L h-1 kg-1) 1299.3 ± 522.9 3622.0 ± 589.6 2.8-fold decrease Vdobs (L kg-1) 5158.6 ± 3397.4 13154.8 ± 2483.3 2.6-fold decrease t1/2 (h) 2.6 ± 0.8 2.6 ± 0.6 None 36 Next experiments • Still have untapped potential in the liver samples • They were flash frozen so cannot do hepatocyte/transporter work • Plan to make microsomes and measure CYP/UGT activities to explore the inhibition independently – CYP2C8/CYP3A4 – UGT1A1/1A3 (more complicated) • Transporter work will need to be done in vitro – Clear evidence of uptake interactions – Efflux transporter issues may also be involved 37 Conclusions • Individual CYP inhibition effects can be modeled well in vitro; repaglinide does seem to have CYP2C8/2C22-specific metabolites but not necessarily as expected • More complete systems have both advantages and disadvantages • In the case of gemfibrozil and repaglinide, transporter inhibition appeared to be much more involved in PK changes than CYP inhibition – Still some work to be done • Rodent utility in transporter studies needs further study 38 Acknowledgements • XenoTech – – – – – – – Phyllis Yerino Forrest Stanley Dr. Sylvie Kandel Seema Muranjan Chandra Kollu Dr. David Buckley Brian Ogilvie • Xenometrics – Dr. Kristin Russell – Tom Haymaker 39 Thank you Questions? Joanna Barbara, Ph.D. Division Director, Analytical Services XenoTech, LLC jbarbara@xenotechllc.com 40