Thompson.Evolutionary divergence Final
advertisement

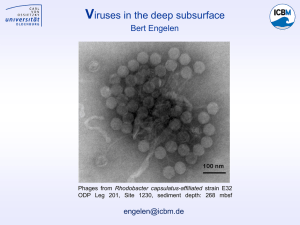
Evolutionary divergence of Plasmids and Phages detected within Clostridium taeniosporum. Thompson, Jose §, Blinkova, A. ¥, Hunicke-Smith, S. ¥, Satterwhite, E. ¥, Tucker, H. ¥, Walker, J.R. ¥, Cambridge, J. M. ¥, León, A. J. §, and Ginés-Candelaria, E.§ §Miami Dade College-Wolfson Campus, Department of Natural Sciences, Health & Wellness, Miami, FL 33132 ¥University of Texas at Austin, Austin TX 78712 ABSTRACT RESULTS Clostridium taeniosporum is a Gram-positive, nonpathogenic, and anaerobic bacterium that was isolated from Crimean lake silt. The organism is approximately 98% homologous in rDNA to toxigenic Clostridium botilinum Group II strains. It is also unique in that it displays characteristic ribbon-like endospore appendages (Lyer AV, 2008). Clostridium taeniosporum’s genome is approximately 3, 452.763 Kbs in length, arranged by sequencing and assembled into 18 scaffolds of various sizes. Upon further analysis of these scaffolds, various putative phages were identified that may have integrated through various evolutionary processes as a result of horizontal gene transfer. To study the evolutionary divergence of phages detected within the Firmicutes, we utilized CLUSTALW multi-wise nucleotide alignment algorithms, under the software platform Geneious Pro v6.1.5 to construct a phylogenetic tree of Bacillus and Clostridia phage genome sequences identified in the NCBI Genbank. These included the phage genomes detected (using the PHAST algorithm, Moreno, Cambridge, personal communication) in Clostridium taeniosporum (six putative bacteriophages, namely phages phiC2, phi2626, phi3636, Geobacillus virus E2, Bacillus Wbeta, and Cp1). Studying the evolutionary divergence of phages detected within Firmicutes may possibly reveal common genome integration mechanisms, uncover close evolutionary relationships, and may provide further insights into the acquisition of cryptic virulence determinants within the group. RESULTS The comparison of phage sequences identified in Clostridium taeniosporum using the PHAST tool, with those detected through CLUSTALW multi-wise alignment, from our collaborators at University Texas exhibited a very high percent homology. Further Comparison of the consensus sequences with those found on he NCBI Genbank revealed that phages phiC2 - 43.8%, Phi3636 - 52.9%, phi2626 - 53.7%, Cp1(Unkown), Wbeta (Unknown) and Geobacillus virus E2 - 40.4% might be incomplete. As illustrated in the phylogenetic tree (Figure 2) phages phiC2, Phi3636, phi2626, Cp1 and Geobacillus virus E2 are clustered in the center of the tree. Preliminary analysis of the location of these show they are more closely related to phages that integrated into Lactobacillus and Clostridium than to the Bacillus spp. DISCUSSION/CONCLUSION We embarked on a study of the evolutionary divergence of the Firmicutes (namely, Bacillus and Clostridium), by studying phages that integrated into C. taeniosporum. With the Bioinformatics software Geneious Pro v6.1.5, we annotated phage sequences that upon further examination of the sequence and its flanking regions using Phage Alignment Tool (PHAST), resulted in the detection of integrated phage genomes within C. taeniosporum. These results were also expanded by our collaborators at University of Texas who identified three additional phage genomes and verified our three isolates, again by using the PHAST tool. Along with various algorithms such as the Jukes-Cantor distance matrix, we were able to provide preliminary aspects of this study. Analysis of Phylogenetic pair-wise alignments of our detected phage genomes found in C. taeniosporum and those sequences published in NCBI Genbank, indicated that most of the phages detected in C. taeniosporum seem to represent incomplete genomes (data not shown). What can be extrapolated from this information is that the six phages most likely may have undergone evolutionary changes/adaptations within the host after integration. The phylogenetic tree of the concatenated multi-wise alignments illustrated a very interesting result. The six phages found within C. taeniosporum are clustered in the of this tree, grouped with phages that integrated into Clostridium and Lactobacillus. Preliminary interpretation of this data indicates that phages, which integrate into Clostridium and Lactobacillus bacteria, may undergo the similar evolutionary changes or site substitutions. Further study of this preliminary work with the Firmicutes, may reveal closer evolutionary relationships, common genome integration mechanisms and insights into the acquisition of cryptic virulence determinants within Clostridium taeniosporum. MATERIALS & METHODS In order to study the evolutionary divergence of Clostridium taeniosporum, we identified the phages that had integrated within this bacterium using PHAST. These sequences were then compared to those identified by our collaborators at University of Texas and those recorded in the NCBI Genbank. The six identified putative phage sequences (phiC2, phi2626, phi3636, Geobacillus virus E2, Bacillus Wbeta, and Cp1) were used to build a CLUSTALW multi-wise nucleotide alignment with the genome sequences of phages that integrated into Firmicutes through the software Geneious Pro v6.1.5. We chose first to compare those that integrate into Bacillus, after which those that integrate into Clostridium. The alignments represent concatenates of various multi-wise alignments considering the large number of sequences involved. These results represent preliminary analyses, which were published using the JukesCantor distance matrix tree-building algorithm in the software Geneious Pro v6.1.5. RESULTS REFERENCES/ACKNOWLEDGEMENTS You Zhou, Yongjie Liang, Karlene Lynch, Jonathan J. Dennis, David S. Wishart “PHAST: A Fast Phage Search Tool” Nucl. Acids Res. (2011) 39(suppl 2): W347W352 [doi:10.1093/nar/gkr485][PMID:21672955] Geneious version v6.1.5 created by Biomatters. Available from http://www.geneious.com/ We would like to thank the National Science Foundation Advanced Technological Education Program NSF ATE DUE 0802508 “The Biotechnbology Research Learning Collaborative” (BRLC) and the US Department of Education, HIS-STEM Program P031C110190 STEM-TRACK for their support of this research project. Figure 2. CLUSTALW multi-wise alignments of phages identified within Bacillus and Clostridium illustrated in a Phylogenetic tree produced by the Jukes-Cantor algorithm within Geneious Pro v6.1.5. Figure 1. Detail prophage view (linear map) of GeoBacillus Virus E2 as described in PHAST.