分子进化分析等
advertisement

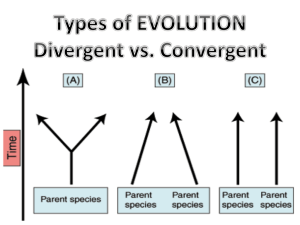
生物信息学 一、分子进化分析 (Molecular Evolution Analysis) 二、表达序列分析 (Analysis of Expressed Sequences) 三、基因芯片数据分析 (Analysis of Microarray Data) 四、SNP分型技术与数据资源 (SNP Genotyping Technologies and Resources) 生物信息学 一、分子进化分析 (Molecular Evolution Analysis) 一、分子进化分析 1. 分子进化分析介绍 2. 系统发育树重建方法 3. Phylip软件包在分子进化分析中的应用 1. 分子进化分析介绍 进化:是一种不断改进的过程。 “每个生物每时每刻都在为生存进行反复的斗争, 如果在复杂多变的生存条件下该生物仍然能够不断 改进自己,那么其将有较大的生存可能性,并被自 然选择所保留。被自然选择保留下来的物种都倾向 于繁殖其已经被改进的新的生命形式” -----《特种起源》 18世纪之前,神创论和物种不变论。 18世纪,相信物种是变化的。拉马克用环境作用的影响、 器官的用进废退和获得性的遗传等原理解释生物进化过程, 创立了第一个比较严整的进化理论。 1859年达尔文发表《物种起源》,论证了地球上现存的生 物都由共同祖先发展而来,并提出自然选择学说以说明进化 的原因,从而创立了科学的进化理论。 20世纪30年代,综合进化论,综合了细胞遗传学、群体遗 传学以及古生物学等学科的成就,进一步发展了进化理论。 20世纪60年代末,分子进化中性学说,认为种内和种间大 多数可见差异是适合度很小的随机突变的固定所决定的。 生物学家:We have a dream… Tree of Life: 重建所有生物的进化历史并以系统 树的形式加以描述 梦想走进现实:How? 1. 最理想的方法:化石!—— 零散、不完整 2. 比较形态学和比较生理学:确定大致的进化框 架 —— 细节存很多的争议 梦想走进现实:How? 3. 第三种方案:分子进化 1964年,Pauling等提出分子进化理论: (1) 生命起源:有机分子由简单向复杂演变 (2) 生物进化:构成生物体的生物大分子如蛋白质、 核酸的演变。 基本假设:核苷酸和氨基酸序列中含有生物进化历 史的全部信息 意义:分子进化的研究可以为生物进化过程提供佐 证,为深入研究进化机制提供重要依据。 分子进化的模式 (1) DNA突变的模式:替代,插入,缺失,倒位 (2) 核苷酸替代:转换 (Transition) & 颠换 (Transversion) (3) 基因复制:多基因家族的产生以及伪基因的 产生 A. 单个基因复制 – 重组或者逆转录 B. 染色体片断复制 C. 基因组复制 (1) DNA突变的模式 插入 替代 酪氨酸 酪氨酸 缺失 倒位 亮氨酸 半胱氨酸 苯丙氨酸 甲硫氨酸 (2) 核苷酸替代:转换 & 颠换 A: 腺嘌呤 C: 胞嘧啶 G: 鸟嘌呤 T: 胸腺嘧啶 转换:嘌呤被嘌呤替 代,或者嘧啶被嘧啶 替代 颠换:嘌呤被嘧啶替 代,或者嘧啶被嘌呤 替代 (3) 基因复制:A. 单个基因复制 重组 逆转录 (3) 基因复制: B. 染色体片段复制 1. 缺失—染色体失去了片段 2. 重复—染色体增加了片段 3. 易位—非同源染色体间相 互交换染色体片段,造成 染色体间的重新排列 4. 倒立—染色体片段作 180°的颠倒,造成染色 体内的重新排列。 (3) 基因复制: C. 基因组复制 K. Waltii (克鲁雄酵母) S. Cerevisiae (酿酒酵母) 克鲁雄酵母 中的同源基 因数量与酿 酒酵母相比 为1:2 分子进化研究的目的 (1) 从物种的一些分子特性出发,构建系统发育树, 进而了解物种之间的生物系统发生的关系 —— tree of life,物种分类 (2) 大分子功能与结构的分析:同一家族的大分子, 具有相似的三级结构及生化功能,通过序列同源 性分析,进行大分子功能预测 (3) 进化速率分析:例如,HIV的高突变性,哪些位 点易发生突变? (1) Tree of Life: 16S rRNA 真菌 古生菌 真核生物 Out of Africa 随着距非洲距离越来越长, 遗传多样性的衰退程度,正 好沿着人类早期迁徙的路线 慢慢增大。 人类迁移的路线 53个人的线粒体基因组(16,587bp) 非洲人相对其他大陆上的 人类在基因上极为多样化 (2) 同源性分析->功能相似性 Paralog (旁系同源物):两个基因在同一物种中, 通过至少一次基因复制的事件而产生 Ortholog (直系同源物):两个基因在不同物种中。 源于不同物种的最近的共同祖先的两个基因,或者 两个物种中的同一基因 Xenolog (异同源物):由某一个水平基因转移事件 而得到的同源序列 Convergent evolution(趋同进化): 通过不同的 进化途径获得相似的功能 旁系同源物vs.直系同源物 paralogs orthologs 异同源物 由某一个水平基因转移事件而得到的同源序列 趋同进化 不同的生物,甚至在进化上相距甚远的生物,如果生活在 条件相同的环境中,在同样选择压的作用下,有可能产生 功能相同或十分相似的形态结构,以适应相同的条件。 鲸、海豚等和鱼类的亲缘关系很远,前者是哺乳类,后者 是鱼类,但形状相似。 鸟类与蝙蝠 基因的趋同进化 通过不同的进化途径获得保守/相似的功能 (3) HIV protease: 高突变性 Ka/Ks >> 1, 强的正选择压力,具有很高的可突变 性 (Ka: 异义突变率,Ks:同义突变率) 系统发育树(Phylogenetic tree) 生命三界: 细菌(Eubacteria) 古细菌(Archaebacteria) 真核(Eukaryotes) 基于16S/18S核糖体RNA序列比对得到的古细菌系统发育树 系统发育树(Phylogenetic tree) 对一组实际对象的世系关系的描述(如基因,物种等)。 末端分支 末端 物种 顶端 叶子 用一种类似树状分支的图 形来概括各种(类)生物之 间的亲缘关系 中间节点 中间枝条 通过比较生物大分子序列 差异的数值构建的系统树称 为分子系统树 节点 根 一个系统发育树 系统发育树的术语 分支/世系 末端节点 A B C D 祖先节点/ 树根 内部节点/分歧点,该 分支可能的祖先结点 E 代表最终分 类,可以是 物种,群体 ,或者蛋白 质、DNA、 RNA分子等 系统发育树的术语 系统发育树是一种二叉树。由一系列节点(nodes) 和分支(branches )组成,其中每个节点代表一 个分类单元(物种或序列),而节点之间的连线 代表物种之间的进化关系。 树的节点又分为外部节点(terminal node)和内 部节点(internal node)。外部节点代表实际观察 到的分类单元。内部节点又称为分支点,代表分 类单元进化历程中的祖先。 系统发育树的种类: 有根树、无根树 理论上,一个DNA序列在物种形成或基因复制时, 分裂成两个子序列,因此系统发育树一般是二叉的。 一般考虑二叉的树结构:二叉树 拓扑结构: 有根树:反映时间 顺序 无根树:反映距离 a b c d a c b d 系统发育树的种类: 有根树、无根树 archaea eukaryote archaea 无根树 archaea eukaryote eukaryote eukaryote Rooted by outgroup bacteria outgroup archaea 外围支 Monophyletic group(单源支) archaea archaea eukaryote 有根树 eukaryote root eukaryote eukaryote Monophyletic group 选择外围支 (Outgroup) 1. 选择一个或多个已知与分析序列关系较远的序列 作为外围支 2. 外围支可以辅助定位树根 3. 外围支条件:序列必须与剩余序列关系较近,但 外围支序列与其他序列间的差异必须比其他序列 之间的差异更显著 系统发育树的种类:基因树、物种树 基因树: 由来自各个物种的一个基因构建的系统发育树(不完全等 同于物种树),表示基因分离的时间。 物种树: 代表一个物种或群体进化历史的系统发育树,表示两个物 种分歧的时间。 Gene tree a A b B c D Species tree 进化分支图,进化树 Bacterium 1 Bacterium 2 Bacterium 3 Eukaryote 1 Eukaryote 2 进化分支图:只有 分支信息,无支长 信息。 Eukaryote 3 Eukaryote 4 Bacterium 1 Bacterium 2 Bacterium 3 Eukaryote 1 进化树:有分支和 支长信息。 Eukaryote 2 Eukaryote 3 Eukaryote 4 系统发生树性质 如果是一棵有根树,则树根代表在进化历史上 是最早的、并且与其它所有分类单元都有联系的分 类单元。 如果找不到可以作为树根的单元,则系统发生 树是无根树。 从根节点出发到任何一个节点的路径指明进化 时间或者进化距离。 系统发生树性质 对于给定的分类单元数,有很多棵可能的系统 发生树,但是只有一棵树是正确的。 系统发生分析的目标 —— 寻找这棵正确的树 分子进化研究的基础 假设:核苷酸和氨基酸序列中含有生物进化历史的 全部信息。 理论:在各种不同的发育谱系及足够大的进化时间 尺度中,许多序列的进化速率几乎是恒定不变的。 (分子钟理论, 1965 ) 虽然很多时候仍然存在争议,但是分子进化确实能 阐述一些生物系统发生的内在规律。 分子钟理论 从一个分歧数据可以推测其他 序 列 分 歧 度 y x 分歧时间 分子钟与线性树 1. 物种分化时间的推断:最理想应该是化石证据 2. 由于化石证据的不足,可以采用分子数据推测物 种的分化时间 3. 给定一个进化树, 已知: A. 分支长度 B. 其中一个分歧点的分化时间 推测所有分歧点的分化时间: 突变的速率恒定 实际数据中 1. 同义替代与非同义替代的速率不同 2. 不同的基因/蛋白质,其进化的速率不同 3. 对于特定的基因,具有一定的、恒定的进 化速率 基因同义替代与非同义替代的速率 速率恒定的证据:血色素 分子钟假设 1. 序列之间的遗传差异的数量是自分化以来 的时间的函数 2. 分子变化的速率相当稳定,可以用来预测 分化的时间 分子钟: 进化时间的估计 1. 遗传距离d的计算: A. 氨基酸序列:p-距离,d-距离,Γ-距离 B. DNA序列:Jukes-Cantor距离,Kimura 距离 2. 物种分歧点:使用考古数据确定共有祖先,确定 分化时间T 3. 计算分子的分化/进化的速率:r=d/2T 4. 对新的序列,计算分化时间: Tnew=dnew/2r 物种分化时间:化石证据 1. 灵长目-啮齿动物: ~80 Myr ago 2. 哺乳动物-鸟类: ~310 Myr ago 3. 哺乳动物-两栖类: ~350 Myr ago 4. 四肢动物-硬骨鱼: ~430 Myr ago 5. 脊椎动物-果蝇 (昆虫): ~830 Myr ago 一、分子进化分析 1. 分子进化分析介绍 2. 系统发育树重建方法 3. Phylip软件包在分子进化分析中的应用 系统发育树重建分析步骤 多序列比对(自动比对,手工比对) 建立取代模型(建树方法) 建立进化树 进化树评估 系统发育树的构建 1. 系统发育树:分子进化树/分子进化分析 2. 通过进化树的构建,分析分子之间的起源关系, 预测分子的功能 3. 建树方法: A. 最大简约法 (Maximum Parsimony) B. 距离法 (distance-based methods) C. 最大似然性法 (Maximum Likelihood) A. 最大简约法(MP) 最大简约法(maximum parsimony,MP)最早源 于形态性状研究,现在已经推广到分子序列的进 化分析中。 最大简约法的理论基础是奥卡姆(Ockham) 原则,这个原则认为:解释一个过程的最好理论 是所需假设数目最少的那一个。对所有可能的拓 扑结构进行计算,计算出所需替代数最小的那个 拓扑结构,作为最优树。 信息位点 (Sites are informative) 1. 必须在至少2个类群中具有相同的序列性状 2. 指那些至少存在2个不同碱基/氨基酸且每个不同 碱基/氨基酸至少出现两次的位点 Position 5, 7, 9为信息位点 1. 基于position 5的三个MP: Tree 1长4,Tree 2 长5,Tree 3长6 2. 计算结果:MP tree的最优结果为tree 1 B. 距离法 又称距离矩阵法,首先通过各个物种之间的比较, 根据一定的假设(进化距离模型)推导得出分类群 之间的进化距离,构建一个进化距离矩阵。进化树 的构建则是基于这个矩阵中的进化距离关系 计算序列的距离,建立距离矩阵 通过距离矩阵建进化树 简单的距离矩阵 通过矩阵建树的方法 由进化距离构建进化树的方法有很多,常见有: 1.Fitch-Margoliash Method(FM法) 2. Neighbor-Joining Method (NJ法/邻接法): 求最短 支长,最通用的距离方法 3. Neighbors Relaton Method(邻居关系法) 4.Unweighted Pair Group Method (UPGMA法) C. 最大似然法(ML) 选取一个特定的替代模型来分析给定的一组序 列数据,使得获得的每一个拓扑结构的似然率都 为最大值,然后再挑出其中似然率最大的拓扑结 构作为最优树。 位置1 位置2 位置3 … SUM 拓扑树A L(A1) L(A2) L(A3) … Asum 拓扑树B L(B1) L(B2) L(B3) … Bsum 拓扑树C L(C2) L(C3) L(C3) … Csum … … … … … … 似然值最大,即SUM最大的拓扑树则为最优树。 构建进化树的一般原则 1. 可靠的待分析数据 2. 准确的多序列比对 3. 选择合适的建树方法: A. 序列相似程度高,MP(最大简约法) B. 序列相似程度较低,ML(最大似然法) C. 序列相似程度太低,无意义 4. 一般采用两种及以上方法构建进化树,无显著区 别可接受 构建进化树的一般原则 进化树的可靠性分析 进化树的可靠性分析: 自展法 (Bootstrap Method) (统计方法) 。 从排列的多序列中随机有放回的抽取某一列, 构成相同长度的新的排列序列 重复上面的过程,得到多组新的序列 对这些新的序列进行建树,再观察这些树与原 始树是否有差异,以此评价建树的可靠性 常用分子进化与 系统发育分析的软件 软件名称 PHYLIP PAUP 网址 说明 It includes programs to carry out parsimony, distance matrix methods, maximum likelihood, and other methods on a variety of http://evolution.gs.washingt types of data, including DNA and RNA on.edu/phylip.html sequences, protein sequences, restriction sites, 0/1 discrete characters data, gene frequencies, continuous characters and distance matrices. http://paup.csit.fsu.edu/ It includes parsimony, distance matrix, invariants, and maximum likelihood methods and many indices and statistical tests. http://phylogeny.arizona.ed Tree of Life u/tree/program/program.htm Arizona大学开发的软件 l MEGA 美国宾州州立大学Masatoshi Nei开发 http://www.megasoftware.n (It carries out parsimony, distance matrix et and likelihood methods for molecular data.) 软件名称 网址 说明 MOLPHY http://www.ism.ac.jp/soft 日本国立统计数理研究所开发。(Carrying out maximum likelihood inference of phylogenies ware/ismlib/softother.e.ht for either nucleotide sequences or protein ml#molphy sequences.) PAML http://abacus.gene.ucl.ac. programs for the ML analysis of nucleotide or uk/software/paml.html protein sequences.) 英国伦敦学院Z. H. YANG开发。(A package of PUZZLE TreeView phylogeny ftp://fx.zi.biologie.uni- 应用Quarter puzzling方法(一种最大简约 muenchen.de/pub/puzzle 法)构建系统发育树 A program for displaying trees on Apple http://taxonomy.zoology. Macs and Windows PCs. It can draw rooted gla.ac.uk/rod/treeview.ht and unrooted trees, display bootstrap values, ml and supports the native font and graphics file formats of both Macs and PCs. http://www.ebi.ac.uk/phy EBI的系统发育树分析软件 logeny.html 一、分子进化分析 1. 分子进化分析介绍 2. 系统发育树重建方法 3. Phylip软件包在分子进化分析中的应用 常见的分子进化分析程序 1. Phylip 由华盛顿大学遗传学系开发,是一个免费的系统发 育分析软件包(版本3.69),可以通过以下地址下载。 http://evolution.gs.washington.edu/phylip.html 2. PAUP* 最早是在苹果机上开发的具有菜单界面的进化分析 软件,早先版本只有MP法,后续版本已经包括距 离法和ML法,现今有mac,win,linux等多种版本, 该软件不是免费软件。 http://evolution.gs.wash ington.edu/phylip.html Phylip软件包介绍 Phylip包含了35个独立的程序,这些独立的程序 都实现特定的功能,这些程序基本上包括了系统 发生分析的所有方面。 Phylip有多种不同平台的版本(包括Windows, Macintosh,DOS,Linux,Unix和OpenVMX)。 Phylip软件包介绍 Phylip是目前最广泛使用的系统发生分析程序,主 要包括一下几个程序组:分子序列组,距离矩阵 组,基因频率组,离散字符组,进化树绘制组。 分子序列组: 1.蛋白质序列:protpars,proml,promlk, protdist 2.核酸序列:dnapenny,dnapars, dnamove,dnaml,dnamlk, dnainvar,dnadist,dnacomp Phylip软件包介绍 距离矩阵组:Fitch,kitsch,neighbor 基因频率组:Gendist,contml 离散字符组: Pars,mix,move,penny,dollop, dolmove,dolpenny,clique,factor 进化树绘制组:drawtree,drawgram 其他:restdist,restml,seqboot,contrast treedist,consense,retree Phylip软件包的文档 Phylip软件包的文档是非常详细的,对于每个独立 的程序,都有一个独立的文档,详细的介绍了该 程序的使用及其说明。 此外,Phylip软件包还包括程序的源代码(c语 言)。 Phylip软件包的应用 1. 根据分析数据,选择适当的程序 如果分析的是DNA数据,就在核酸序列分析类中 选择程序(dnapenny,dnapars, dnamove,dnaml,dnamlk, dnainvar,dnadist,dnacomp ) 如果分析的是离散数据,如突变位点数据,就在 离散字符组里面选择程序。 Phylip软件包的应用 2. 选择适当的分析方法 如分析的是DNA数据,可以选择简约法 (DNAPARS),似然法(DNAML, DNAMLK),距离法等(DNADIST)。。。 3. 进行分析 选择好程序后,执行,读入分析数据,选择适当 的参数,进行分析,结果自动保存为outfile, outtree。 Phylip软件包的应用 Outfile是一个记录文件,记录了分析的过程和 结果,可以直接用文本编辑器(如写字板)打开。 outtree是分析结果的树文件,可以用phylip提供 的绘树程序打开查看,也可以用其他的程序来打开, 如treeview。 生物信息学 二、表达序列标签(EST) 测定及分析 二、表达序列标签(EST)测定及分析 1. EST的概念 2. EST的应用 3. EST序列测定及分析过程 EST的概念 EST是指通过对cDNA 文库随机挑取的克隆进行 大规模测序所获得的cDNA 的5′或3′端序列,长度 一般为60 ~ 500 bp. EST 是基因的“窗口”,可代表生物体某种组织 某一时间的一个表达基因,故被称之为“表达序列 标记” (Expressed Sequence Tag, EST) EST的来源 上世纪80年代,对肾上腺素受体基因进行分离和 测序,花了10年时间才完成了基因的克隆与测序。 上世纪80年代,对cDNA序列进行大规模测序的 想法就曾提出,但反对者认为cDNA序列缺少重要的 基因调控区域的信息。 EST技术应用的首次报道是Adams(1991)等从三 种人脑组织cDNA文库随机挑取609个克隆进行测序, 得到一组人脑组织的EST,分析结果表明其中36个 代表已知基因,337个代表未知基因。 NIH 运用自动化测序技术, 大规模生产EST 序列。 总EST条目 (million) dbEST中数据量的增长 20 18 16 14 12 10 8 6 4 2 0 1993 1995 1997 1999 2001 时间(年) ● 1993年前ESTs数据收录于GenBank, EBI和DDBJ。 ● 1993年NCBI建立了一个专门的EST数据库dbEST来 保存和收集所有的EST数据。 二、表达序列标签(EST)测定及分析 1. EST的概念 2. EST的应用 3. EST序列测定及分析过程 1. ESTs与新基因识别 发现新基因是基因组研究的热点, 使用生物信息学方法预测 新基因是后基因组时代必不可少的方法。 利用对某一特异组织或某一生长发育阶段的cDNA 文库, 进 行随机部分测序所得的EST s, 作为查询项在dbEST 中进行同 源查找, 同时将由EST s 序列按密码子推出的氨基酸序列作为 查询项在蛋白质信息资源数据库中进行同源查找。 如果该EST s 序列在以上数据库中存在同源序列, 可对该 EST s 所代表基因的功能进行分析及鉴定。如果不存在同源序 列, 则该EST s 所代表的基因有可能是新基因。 2. ESTs与遗传学图谱的构建 物理图谱、遗传图谱和转录图谱是基因组计划要获得的三 种遗传学图谱。 转录图谱为染色体DNA 某一区段内,所有可转录序列的分 布图,ESTs作为转录基因的产物,可直接用于构建转录图谱。 由于ESTs具有很高的多态性可用作分子标记,用于建立遗 传连锁图谱。 建染色体物理图谱需要大量的单拷贝序列标记位点(STS) 作为界标,由于大多数基因是单拷贝的,因此ESTs可以充当 STS构建物理图谱。 2. ESTs与遗传学图谱的构建 序列标签位点(sequence-tagged sites, STS)是从人类基因组 中随机选择出来的长度在200-300bp左右的经PCR检测的基因 组中唯一的一段序列。 来自mRNA的3’非翻译区的ESTs更适合做为STSs,用于基因 图谱的绘制。优点: ● 由于没有内含子的存在,因此在cDNA及基因组模板中其 PCR产物的大小相同。 ● 与编码区具有很强的保守性不同,3’UTRs序列的保守性 较差,因此很容易将单个基因与编码序列关系非常紧密的相 似基因家族成员分开。 3. ESTs与基因预测 由于EST来源于cDNA,因此每一条EST均代表 了文库建立时所采样品特定发育时期和生理状态下 的一个基因的部分序列。 大于90%的已经注释的基因都能在EST库中检 测到。 ESTs可以做为其它基因预测算法的补充。 4. ESTs与单核苷酸多态性(SNPs) 通过对ESTs重叠群组装,对大量重复的ESTs进行序列比 较,可以从ESTs数据库中筛选另一种以测序为核心的分子标 记SNPs。来自不同个体的ESTs可用于发现基因组中转录区 域存在的SNPs。 注意区别真正的SNPs和由于测序错误而引起的本身不存 在的SNPs。解决这一问题可以通过: ● 提高ESTs分析的准确性。 ● 对所发现的SNPs进行实验验证。 5. ESTs与基因的差异表达 某一时期基因表达的数量通常占全部基因的15%,细胞的分 化由基因特异性的时空表达决定。利用未经标准化和差减杂交 的cDNA文库EST可以分析特定组织的基因表达谱。 近年来对基因差异表达研究的方法有ESTs法、差减杂交法 和mRNA 差异显示技术。其中以ESTs 法稳定性最高, 分析规 模最大。 心脏发育过程基因的差异表达 5. ESTs与基因的差异表达 癌症基因组解析计划 (Cancer Genome Anatomy Project , CGAP) 为研究癌症的分子机理,美国国家癌症研究所NCI 的CGAP计划,构建了很多正常的或是癌症前期 的和癌症后期的组织的cDNA文库,并进行了大规 模的EST测序。CGAP网站提供了多种工具用以分 析不同文库间基因表达的差异,如: ● Digital Gene Expression Displayer (DGED) ● cDNA xProfiler 6. ESTs与DNA芯片的制备 DNA 芯片是指将许多许多特定的DNA 寡核苷酸或DNA 片 段(包括cDNA ) 固定在芯片的每个预先设置的区域内, 将 待测样本标记后同芯片进行杂交, 通过杂交信息的分析来 检测基因的功能和基因组研究的分析系统。 ESTs 是用于制备DNA 芯片的很好基因资源。由于EST s 直接来源于cDNA , 因此EST s 文库可代表cDNA 文库用 于制备DNA 芯片所需的探针库。 绿色: 基因表达↓ 红色: 基因表达↑ 黄色: 基因表达相当 7. ESTs与基因表达系列分析 基因表达系列分析(Serial Analysis of Gene Expression, SA GE) 技术, 能同时对上千个转录物进行研究,是一种用于定量及高通量基 因表达分析的实验方法。 SAGE的原理: (1)一个9-14 碱基的短核苷酸序列标签包含有足够的信息,能够唯一 确认一种转录物。一个9 碱基顺序能够分辨262,144 个不同的转录物, 而人类基因组估计仅能编码80,000种转录物,所以理论上每一个9 碱 基标签能够代表一种转录物的特征序列。 (2)将短片段标签相互连接形成长的DNA 分子,对该克隆进行测序得 到大量连续的单个标签,可对数以千计的mRNA 转录本进行分析。 (3)特定的序列标签的出现次数就反应了对应的基因的表达丰度。 基因表达系列分析 (SAGE) 技术流程 反转录 酶切 连接 测序 实 验 步 骤 较 长 要 求 较 高 单条测序=对30-40条EST测序 分析 由于采样量大大提高,可对低表达基因进行分析: 基因表达量分析、寻找新基因等等 ESTs数据的不足 ESTs很短,没有给出完整的表达序列。 低丰度表达基因不易获得。 由于只是一轮测序结果,出错率达2%-5%。 有时有载体序列和核外mRNA来源的cDNA污染或 是基因组DNA的污染。 有时出现镶嵌克隆。 序列的冗余,导致所需要处理的数据量很大。 二、表达序列标签(EST)测定及分析 1. EST的概念 2. EST的应用 3. EST序列测定及分析过程 EST技术流程 体内:翻译 体外研究:反转录 连接,转化 大数据量分析理念已经形成 文库构建技术已经成熟 测序成本已经大大降低 A. cDNA文库构建 ◆ 非标准化的cDNA文库的构建。 可用于基因表达量的分析 ◆ 经标准化或扣除杂交处理的cDNA文库。 富集表达丰度较低的基因 Oligo(dT) cDNA文库: 在 cDNA 的合成过程中加入高浓度的 Oligo (dT)引物, Oligo(dT)引物与 mRNA 的 3’末端的 poly(A)配 对,引导反转录酶以 mRNA 为模板合成第一链 cDNA 。应用普遍。 随机引物cDNA文库:随机引物引导的 cDNA 合成是采用 6-10 个随机 碱基的寡核苷酸短片段来锚定 mRNA 并作为反转录的起点。 cDNA文库的构建 B. 序列测定及数据分析 随机挑取克隆进行5’或3’端测序 序列前处理 聚类和拼接 基因注释及功能分类 后续分析 1. 测序方向的选择 ◆ 5’端 5’上游非翻译区较短且含有较多的调控信息。一般在寻找新基 因或研究基因差异表达时用5’端EST较好,而且从5’端测序有利 于将EST拼接成较长的基因序列。 ◆ 3’端 3’端mRNA有一20-200bp的polyA结构,同时靠近plyA又有 特异性的非编码区,所以从3’端测得EST含有编码的信息较少, 但研究非编码区有品种的特异性,可以作为STS标记. ◆ 两端测序 获得更全面的信息。 2. 序列前处理 (pre-processing) (1) 去除低质量的序列 (2) 应用BLAST、RepeatMasker或Crossmatch遮蔽数据 组中不属于表达的基因的赝象序列(artifactual sequences)。 ●载体序列(ftp://ncbi.nlm.nih.gov/repository/vector) ●重复序列(RepBase,http://www.girinst.org) ●污染序列 (如核糖体RNA、细菌或其它物种的基因组 DNA等) (3) 去除其中的镶嵌克隆:Back-to-back poly(A)+ tails; Linker-to-linker in middle of the sequence. (4) 最后去除长度小于100bp的序列。 3. ESTs的聚类和拼接 聚类的目的就是将来自同一个基因或同一个转录本的具有 重叠部分(overlapping)的ESTs整合至单一的簇(cluster)中。 聚类作用: 产生较长的一致性序列(consensus sequence) ,用于注释。 降低数据的冗余,纠正错误数据。 可以用于检测选择性剪切。 ESTs聚类的数据库主要有三个: UniGene (http://www.ncbi.nlm.nih.gov/UniGene) TIGR Gene Indices (http://www.tigr.org/tdb/tgi/) STACK (http://www.sanbi.ac.za/Dbases.html) 不严格的和严格的聚类 ◆ loose clustering ● 产生的一致性序列比较长 ● 表达基因ESTs数据的覆盖率高 ● 含有同一基因不同的转录形式,如各种选择性剪接体 ● 每一类中可能包含旁系同源基因的转录本 ● 序列的保真度低 ◆ stringent clustering ● 产生的一致性序列比较短 ● 表达基因ESTs数据的覆盖率低 ● 因此所含有的同一基因的不同转录形式少 ● 序列保真度高 Clean Short and Tight TIGR-THC UniGene STACK Long and Loose Cluster(簇)的拼接 利用cDNA克隆的信息和5’、3’端的序列信息,不同的 Cluster可以连接在一起。 常用的拼接软件 ◆ Phrap (http://www.phrap.org/phredphrapconsed.html ) ◆ CAP3 (http://pbil.univ-lyon1.fr/cap3.php) ◆ d2_cluster (http://www.sanbi.ac.za/) 4. 基因注释及功能分类 (1) 注释: ◆ 序列联配 Blastn: search nucleotide databases using a nucleotide query. Blastx: search protein databases using a translated nucleotide query. ◆ 蛋白质功能域搜索(二结构比对) Pfam: The Pfam database is a large collection of protein families, each represented by multiple sequence alignments and hidden Markov models. Interpro: InterPro is an integrated database of predictive protein "signatures" used for the classification and automatic annotation of proteins and genomes. (2) 基因功能分类: ◆ 手工分类 大部分以Adams 1995年的文章中的采用分类体系为标准。 【Adams. MD, et al. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature. 1995 377(6547 Suppl):3-174 】 ◆ 计算机批量处理 利用标准基因词汇体系Gene Ontology,进行近 似的分类。 (http://www.geneontology.org/) http://www.geneontology.org/GO.downloads.annotations.shtml 5. 后续分析 ◆ 比较基因组学分析 ◆ 基因表达谱分析 ◆ 新基因研究 ◆ 基因可变剪切分析 ◆ 实验验证 ► MicroArray ► GeneChip ► RT-PCR ► Northern blotting 生物信息学 三、基因芯片数据分析 (Analysis of Microarray Data) 三、基因芯片数据分析 1. 基因芯片(Microarray)简介 2. 图像处理与数据标准化 3. 基因芯片的数据分析 1. 基因芯片简介 基因芯片 (1987): 固定有寡核苷酸、DNA或cDNA等 的生物芯片。利用这类芯片与标记生物样品进行杂 交,可对样品基因表达谱生物信息进行快速定性和 定量分析。 高通量、点阵以及Northern杂交 同时测定细胞内数千个基因的表达情况 将mRNA反转录成cDNA与芯片上的探针杂交 芯片的体积非常小:微量样品的检测 基因表达情况的定量分析 基因芯片的密度:100-1 million DNA 探针/1cm2 碱基互补 将样品中的DNA/RNA标上荧 光标记,则可以定量检验基 因的表达水平 基因芯片技术的类型 A. 按技术手段、探针类型分类 1. Short oligonucleotide arrays (Affymetrix) 2. cDNA arrays (Brown/Botstein) 3. Long oligo arrays (Agilent) 4. Serial analysis of gene expression (SAGE) B. 按实验要求分类 1. 单通道 (Single Channel): 一次检验一种状态 2. 双通道 (Dual Channel): 差异表达基因的筛选 两类主流的DNA芯片 (1). cDNA microarrays: 将500~5,000bp的cDNA固载 到介质上 (例如玻璃)。Stanford开发设计,通常 为双通道,常用于差异表达基因的筛选。 (2). DNA chips: 将寡核苷酸探针 (20~80-mer) 合成到 芯片上。Affymetrix开发设计,通常为单通道, 一次检验一种状态 。 (1) cDNA microarrays cDNA clones 载玻片 差异表达基因的筛选 Treatment / control Normal / tumor tissue Brain / liver … 荧光标记的靶基因 (2) DNA chips DNA chips的制备:Affymetrix photolitography 探针长度:25 bp 每个基因:22-40个探针 Perfect Match (PM) vs. MisMatch (MM) probes A. 选择硅片、玻璃片、瓷片或聚丙烯膜、尼龙膜等支持物 B. 采用光导化学合成和照相平板印刷技术在硅片等表面合成寡核苷酸探 针; 或者通过液相化学合成寡核苷酸链探针,或PCR技术扩增基因序列, 由阵列复制器,或阵列机及电脑控制的机器人,将不同探针样品定量点 样于带正电荷的尼龙膜或硅片等相应位置上 C. 紫外线交联固定后即得到DNA微阵列或芯片 三、基因芯片数据分析 1. 基因芯片(Microarray)简介 2. 图像处理与数据标准化 3. 基因芯片的数据分析 2. 图像处理与数据标准化 单通道基因芯片 white (very high) red (high) Yellow (a little high) green (medium) blue (low) black (no) 图像处理 1. 2. 3. 4. 栅格化:确定点的位置 图象分割 (Segmentation):将点从背景中分离出来。 抽提亮度:各个像素亮度的平均值 (mean)或中位数 (median) 背景校正:局部或全局 植根区域生长法(SRG) Fixed Circle 基因表达量的定量 对于每个点,可以计算 Red intensity = Rfg - Rbg fg = foreground, bg = background, and Green intensity = Gfg - Gbg and combine them in the log (base 2) ratio Log2( Red intensity / Green intensity) Green intensity (medium): ~1 Microarray: 误差的来源 log signal intensity 1. 图像分析 2. 扫描 3. DNA杂交过程 (温度、时间、混合均匀 程度等) 4. 探针的标记 5. RNA的抽提 系统误差 6. 加样 随机误差 7. 其他 log RNA abundance 数据标准化 before after 目的是消除系统偏差引起的高相关性,同时保留由真正生物 学原因引起的基因表达水平高相关性。 三、基因芯片数据分析 1. 基因芯片(Microarray)简介 2. 图像处理与数据标准化 3. 基因芯片的数据分析 3. 基因芯片的数据分析 (1) 差异表达基因的分析 (2) 基因共表达分析 (3) 基因表达数据的聚类 (4) 基因表达数据的分类 (5) Map to GO (6) Gene regulatory network (1) 差异表达基因的分析 差异表达基因的分析: 寻找处理前后表达上调或 者下调的基因 Are the treatments different? 使用标准的统计学方法检验 (t-test or f-test),发 现统计显著性差异表达的基因, 如果处理本身并不显著,则结果无意义 统计学分析 Fold change, 一般2-fold increase or decrease (平行实验的样本较少) p-value (平行实验的样本较多) under-expressed /2 over-expressed /2 P-value: 学生分布 T-test: 学生分布 Excel函数:TTEST(array1,array2,tails,type) Array1为第一个数据集 Array2为第二个数据集 Tails指示分布曲线的尾数。如果 tails = 1,函数 TTEST 使用单尾分布。如果 tails = 2,函数 TTEST 使用双尾分 布 Type为 t 检验的类型 1 成对 2 等方差双样本检验 3 异方差双样本检验 P-value: 学生分布 一般选择双尾分布 异方差双样本检验 Excel函数:=TTEST(B2:D2,E2:G2,2,3) C:对照组;T:实验组 Gene 1 C1 C2 C3 T1 T2 T3 TTEST 1.322 1.676 1.457 3.526 4.234 3.879 0.001988 (2) 基因共表达分析 在N个不同的条件下 (时间序列的芯片数据),考察基 因X和Y的表达是否相似。 Gene 1#是否与Gene 2#、Gene 3#和Gene 4#共表达? 共表达: 正相关:相似的表达谱,可能存在正关联 负相关:相反的表达谱,可能存在负调控 Gene Name T1 T2 T3 T4 T5 T6 Gene 1# 1 2 3 4 5 6 Gene 2# 100 200 300 400 550 610 660 540 430 320 210 Eisen MB, et al., (1998) 1504 215 PNAS 35795:14863-14868 2545 1670 101 Gene 3# Gene 4# 998 基因相关性分析:Pearson相关系数 r ~ [-1, 1] r~ 1,正相关 r~ -1,负相关 Gene 1# Excel函数:=PEARSON(array1,array2) Gene 2# Gene 3# Gene 1# Gene 2# 0.996368 Gene 3# -0.99988 -0.99611 Gene 4# 0.245292 0.254855 -0.2395 结论:Gene 1#与Gene 2#表达正相关,与Gene 3#表达负相关,与Gene 4#无关联 (3) 基因表达数据的聚类 将表达谱相似的基因聚类在一起 发现新的模式 聚类方法: A. Hierarchical clustering B. K-means clustering A. Hierarchical clustering 用树状结构来表征基因表达之间的 相似性/相关性 Cluster Tree OBJECT1 Object 1 2 3 4 5 1 2 2 3 6 4 10 9 4 5 9 OBJECT2 5 8 5 3 Distance matrix OBJECT3 OBJECT4 OBJECT5 0 1 2 3 4 Distances 5 6 Distance Cluster 0 1,2,3,4,5 2 (1, 2), 3, 4, 5 3 (1, 2), 3, (4, 5) 4 (1, 2), (3, 4, 5) 5 (1, 2, 3, 4, 5) B: K-means clustering 对数据进行聚类 必须给定结果分成多 少类 假设该例中,指定为 聚成5类 B: K-means clustering 软件:Cluster 3.0, Michael Eissen, Stanford 最终结果:所有基因 芯片数据被聚成5类 (4) 基因表达数据的分类 根据基因表达的数据将样本分成两类或多类 督导学习 (supervised learning):根据发现的模式 进行预测 应用: 癌症 vs. 正常组织 癌症的亚型、不同阶段 (良性的 vs. 恶性的) 对药物的敏感性 (tamoxifen for breast cancer) (5) Map to GO 通过基因芯片,找到了一批“interesting” 的基 因 生物学功能上是否存在关联? 基因本体(Gene Ontology, GO):主要包括三 个分支: 生物过程、分子功能和细胞组件。 GoToolBox: 基于web的程序,允许从一个基因集 (相对于被检索的参考基因集)识别统计上过表达 或低表达的术语、基因集里对功能相关的基因进 行聚类和检索基因集里共享的注释。 (6) Gene regulatory network 早期观点:表达谱相似的基因可能存在功能上的 关联,可能有相互作用… (直接作用)。 当前的观点:表达谱相似的基因可能具有共同的 调控元件 (基因UTR区域存在共同的Promotor), 能够被同一个上游因子所调控。 生物信息学 四、SNP分型技术与数据资源 (SNP Genotyping Technologies and Resources) 一、SNP简介 人类表型的多样性 人类表型的多样性的原因 1. 生存环境因素的影响 自然条件 文化背景 生活与饮食习惯 社会体制 ……. 2. 自身遗传物质的作用 基因的功能和调节 非编码DNA序列的影响 表观遗传的作用 多态性位点的存在 …… 人类遗传物质的差异:基因多态性 1. 人类遗传物质99.8%是完全一致的 2. 人类遗传物质的差异大约为0.2% 其中: 在核苷酸碱基水平上约占0.08% 比如:SNPs 、点突变、碱基替换、插入或缺失等 在基因组结构水平上约占0.12% 比如:片段的插入、缺失、倒置、移位、互换等 1. 概念 单核苷酸多态性(single nucleotide polymorphism,SNP):主要是指在基因组水平上 由单个核苷酸的变异所引起的DNA序列多态性。 是人类可遗传的变异中最常见的一种,占所有已 知多态性的90%以上。人类基因组平均每1000个碱 基对中就有1个SNP (0.1%),估计其总数可达300万 个甚至更多。 146 Single Nucleotide Polymorphisms (SNPs) 2. 研究意义 SNP作为继RFLP (限制性片段长度多态性)和小卫星DNA 重复序列、微卫星DNA重复序列后的第三代DNA 水平遗 传多态性标记。 在整个基因组中分布密集,与疾病的相关性稳定,是遗 传学研究的新工具,在基因研究和药物设计中被广泛应 用。 广泛应用于基因组分析、生物信息自动化检测、简单和 复杂疾病的遗传研究及全球种族遗传学研究等。 148 3. 特性及优点 (1) SNP数量多,分布广泛:人类有300万以上的 SNPs,遍布于整个人类基因组中。 (2) SNP适于快速、规模化筛查:SNP一般只有两种 碱基组成,由于SNP的二态性,非此即彼,在 基因组筛选中SNPs往往只需+/-的分析,而不用 分析片段的长度。 (3) SNP等位基因频率容易估计:采用混和样本估 算等位基因的频率是种高效快速的策略。 原理:选择参考样本制作标准曲线,然后将待测的混和 样本与标准曲线进行比较,根据所得信号的比例确定混 和样本中各种等位基因的频率。 (4) 易于基因分型:SNPs 的二态性,也有利于对其 进行基因分型。 4. SNP相关的基本概念 等位(allele):同一位置上的每个碱基类型叫做 一个等位,SNP是一种双等位多态。如某些人的染 色体上某个位置的碱基是A,而另些人的染色体的 相同位置上的碱基则是G。 基因型(genotype):一对同源染色体上的两个 等位的组合。 单体型(haplotype):特定染色体区域相邻近 的SNP的组合。 SNP等位、基因型、单体型与Tag SNP 最小等位频率: 群体中,一对SNP等位中出现较 少的等位的频率,以5%为界将SNP分为常见SNP和 罕见SNP。 非同义SNP:指碱基序列的改变可使蛋白质序列 发生改变,从而影响了蛋白质的功能。 同义SNP:SNP所致的编码序列的改变并不影响 其所翻译的蛋白质的序列。 5. 分类: (1) 根据SNP 所处的位置 蛋白编码SNP:位于外显子中,如果它不引起所 编码的氨基酸改变,则称为同义SNP,否则称为 非同义SNP。 非蛋白编码SNP:位于内含子区或基因间区,不 会影响蛋白质序列。 调节SNP(基因周边SNP):位于基因调节区,如 果它影响到基因的表达水平,就会影响到RNA或 蛋白质的产量,从而影响性状。 5. 分类: (2) 根据能否导致遗传后果 致病或成因SNPs (causative SNPs): 可引起疾病 或性状的改变 A. 成因型SNP 可以引起严重的遗传疾病,最经典 的是镰刀型细胞贫血症。 B. 人类消化牛奶的能力,欧洲和非洲一些人有很高 的乳糖酶活性,而世界上约一半成年人不能消化牛 奶,这种差异可由4 个SNPs 单独或共同引起。 代理型SNPs (surrogate SNPs): 不影响表型 二、SNP检测和分型技术 SNP分型(genotyping) 对SNP基因型的检测过程 SNP分型包括内容 对未知SNP进行分析:寻找未知的SNP或确定某 一未知SNP与某遗传病的关系。 对已知SNP进行分析:对不同生物群SNP遗传多 样性检测或在临床上对已知致病基因的遗传病进 行基因诊断。 SNP分型技术 区分SNPs位点的方法 (一) 基于分子杂交的方法 (二) 以SNP影响核酸构象为基础的方法 (三) 基于酶切的方法 (四) 测序方法 (一) 基于分子杂交的方法 原理: 短的核苷酸探针在和互补的目的片段进 行杂交时,完全匹配和有错配两种情况下, 根据杂交复合体稳定性的不同而将SNPs 位点检测出来。 差异越大,检测的特异性就越好。 方法: 1. 等位基因特异寡核苷酸片段分析(allele- specific oligonucleotide, ASO) 通过设计一段15-20bp的寡核昔酸片段, 其中包含了发生突 变的位点, 当与固定在膜上的样品DNA(如PCR产物)杂交时, 由于其中一个碱基的差异会导致Tm值下降5-7.5℃ , 故通过 严格控制杂交的条件, 可鉴定出样品DNA中是否存在SNP。 将各种寡核苷酸片段固定在膜上, 然后用样品DNA来杂交, 这样可同时检测多种突变或多态, 即反向点杂交技术。 2.基因芯片方法 利用寡核苷酸与不同靶序列变异配对的杂交稳定 性的差异, 在一小块硅片上高密度地集成上万乃 至更多的探针形成多重寡核昔酸微阵列, 通过与 目的DNA的杂交荧光显色等方法检测。 将所有SNP全部信息载入DNA芯片, 就可制造 “ 基因扫描仪” , 用来扫描各个个体在基因组成 上的差异。 2. 基因芯片方法 目前常用的SNP芯片单次测量数量达到50万—100 万个SNP,价格降至5000元以下。 3. 分子信标(双分子间杂交) 分子信标是一个U型单链寡核苷酸探针,内部有部 分序列互补配对,在探针的两端分别带有荧光素和 淬灭剂,荧光素和淬灭剂在空间结构上靠得很近, 不会产生荧光。 探针与目标序列完全互补配对后,荧光素与淬灭剂 分开而产生荧光;如果探针与目标序列之间存在错 配碱基,就不会产生荧光。 3. 分子信标(双分子间杂交) 4. TaqMan探针技术 通过检测PCR过程中及之后产生的荧光信号来区分 等位基因类型的SNP。 需要一对TaqMan探针和一条位于待测位点上游的 引物。这对探针序列仅区别于多态性位点,其3′端 连有荧光淬灭剂,5′端分别连有两种不同荧光染料。 PCR中,利用Taq酶5′核酸酶活性降解与目标序列完 全互补的探针,使荧光剂与淬灭剂分离而发出荧光。 若探针与目标序列间存在错配,就会大大减少荧光 的释放量。 Taqman 探针法 5. 动态等位基因特异性杂交(DASH) 将标记有荧光染料的探针与目标序列配对,然后测 定互补双链所发出的荧光强度与反应体系的温度之 间的关系曲线。 当温度达到变性温度时,荧光强度迅速减弱,存在 错配碱基的互补双链的变性温度低于不含错配碱基 的双链。 通过测定互补双链的变性温度来判断互补双链中是 否含有错配碱基。 5. 动态等位基因特异性杂交(DASH) SNP分型技术 区分SNPs位点的方法 (一) 基于分子杂交的方法 (二) 以SNP影响核酸构象为基础的方法 (三) 基于酶切的方法 (四) 测序方法 (二) 以SNP影响核酸构象为基础的方法 1. 变性梯度凝胶电泳(DGGE)和温度梯度凝胶电泳 (TGGE)法 DGGE和TGGE分别通过逐渐增加的化学变性剂线 性浓度梯度和线性温度梯度,把长度相同但只有一 个碱基不同的DNA片段分离。 双链DNA在变性剂(如尿素或甲酰胺)浓度或温度梯 度增高的凝胶中电泳,随变性剂浓度或温度升高, 由于Tm值不同,DNA的某些区域解链,降低其电 泳泳动性,导致迁移率下降。 变性梯度凝胶电泳(DGGE) 温度梯度凝胶电泳(TGGE) 变性梯度凝胶电泳(DGGE) 左,突变型;中,野生型; 右,突变型与野生型。 A .凝胶的变性梯度方向与 电泳方向垂直 B .凝胶的变性梯度方向与 电泳方向平行 2. 单链构象多态性(SSCP) 相同长度的单链DNA,如果碱基顺序不同,甚至 单个碱基不同,就会形成不同的构象,在电泳时泳 动的速度将产生差异。 PCR-SSCP:经PCR 扩增的片段在变性剂或低离 子浓度下经高温处理使之解链并保证在单链状态下, 然后在非变性聚丙稀酰胺凝胶中电泳。 PCR-SSCP 保 持 在 单 链 状 态 非 变 性 凝 胶 电 泳 构 象 不 同 呈 现 多 态 性 PCR-SSCP DNA 单链构象同双链一样,也包括一级结构、 二级结构,其空间结构中最主要的是发夹结构。 发夹结构是决定DNA 单链构象多态性的分子 基础。 SNP分型技术 区分SNPs位点的方法 (一) 基于分子杂交的方法 (二) 以SNP影响核酸构象为基础的方法 (三) 基于酶切的方法 (四) 测序方法 (三) 基于酶切的方法 1. 限制性片段长度多态性(restriction fragment length polymorphism,RFLP) 定义:由于碱基的变异可能导致酶切位点的消失或 新的酶切位点出现,从而引起不同个体在用同一限 制酶切时,DNA片段长度出现差异,这种因内切 酶位点变化所导致的DNA片段长度的差异,称限 制性片段长度多态性(RFLP)。 限制性片段长度多态性(RFLP) 限制性内切酶Ⅱ识别DNA 序列上的的回文结构。 RFLP分析和PCR技术联合应用 理论依据: • PCR扩增目的基因片段 1 • 限制性内切酶酶切目的基因片段 2 • 电泳分离 3 RFLP分析和PCR技术联合应用 RFLP分析和PCR技术联合应用 野 生 型 纯 合 子 突 变 型 纯 合 子 杂 合 子 SNP分型技术 区分SNPs位点的方法 (一) 基于分子杂交的方法 (二) 以SNP影响核酸构象为基础的方法 (三) 基于酶切的方法 (四) 测序方法 (四) 测序方法 1. 直接测序法 测序结果能够完全呈现SNP等位在基因组精确的 排列顺序,提供具有更高信息含量的数据。 第一代测序技术:以Sanger法(双脱氧核苷酸末 端终止法)为代表的第一代测序技术帮助人们完 成了从噬菌体基因组到人类基因组图谱等大量测 序工作。 1. 直接测序法 第二代测序技术:即高通量测序技术。可以在极 短时间内一次完成数十万到数百万条DNA分子的 序列测定。测序通量提升,成本降低。 对基因直接测序获得高通量、全局性的SNP图谱 将很快成为最经济、最准确、应用最广泛的高通 量分型技术。 第二代高通量测序平台 2. 微测序法 定义:基于特异等位基因的高通量SNPs 检测技 术,依赖DNA 聚合酶来分辨碱基多态性位点。 原理:设计一个引物,其3’端结束在突变位点前 一个碱基,然后在反应体系中加入ddNTP,只有 ddNTP 与模板DNA 被检测位点处的核苷三磷酸 互补时,延伸反应才发生,但仅在连接一个碱基 后就停止,通过检测延伸产物来判断SNP类型。 三、SNP数据资源 NIH的dbSNP多态性数据库: www.ncbi.nlm.nih.gov/snp 德国的HGBAS网站的人类SNP数据库: hgbas.cgr.ki.sei 日本建立的JSTSNP数据库: snp.ims.utolkyo.ac.jp NIH的与癌症和肿瘤相关的候选SNP数据库: cgap.nci.nih.gov/GAI UCSC Genome Bioinformatics Site: genome.ucsc.edu 美国Utah大学SNP数据库:www.genome.utah.edu 美国波士顿儿童医院SNP数据库:snpper.chip.org SNP联盟数据库:www.cshl.org 英国Sanger研究所:www.sanger.ac.uk …… 1. NIH的dbSNP多态性数据库 网址:www.ncbi.nlm.nih.gov/snp/ 总数量约为1470万个SNPs。 三大来源: SNP联盟数据库:www.cshl.org 英国Sanger研究所:www.sanger.ac.uk 美国Washington University, St.Louis 设立的参照SNPs>270万个,采用rs+数字编号来表示 dbSNP的挑选方式和不足: 60%的“候选”SNPs是通过统计学方法预测出来的,即通过比较重叠克 隆中的DNA序列痕迹来确定“候选”SNPs。因此,大多数的dbSNP是 频率未知的“候选”SNPs。 dbSNP的质量: 已经证实的Ref_SNPs,大约有240万个 非人类的SNPs,大约有216万个 无法证实的SNPs,大约有184万个 在某一群体中不是多态性的,大约有152万个 在某一群体中频率<20%的,大约有126万个 被证实的其频率>20%的SNPs,大约有63万个 2. SNP生物信息分析 (1) 分析的参数或指标 挑选拟进行分析的基因及DNA序列长度。如5’端上游 5000bp + 整个基因序列 + 3’端下游5000bp,要求包含 两端的非转录区(UTR)。 寻找获取下列的信息 所有SNP的信息: 位置、群体中的频率 标记SNP(Tag_SNP)的情况: 位置、群体中的频率 基因外显子的信息: 位置、方向、大小 转录因子结合位点信息: 名称、位置、数目 甲基化位点CpG的信息: 位置、数目 进化保守区的信息: 名称、位置、数目、大小 参与调节基因转录的序列簇信息: 名称、位置、数目、大小 (比如增强子、沉默子和microRNAs结合域等) (2) 涉及的数据库或网络资源 在http://genome.ucsc.edu 获得SNP、CpG、转录因子结 合位点信息 在http://ecrbrowser.dcode.org 获得进化保守区的信息 在http://www.ensembl.org/index.html 获得基因外显子的 信息 在http://www.hapmap.org 获得标记SNP(Tag_SNP)、单 倍型的信息 在http://genome.perlegen.com/browser/index_v2.html 获得SNP、单倍型的信息 在http://zlab.bu.edu/cluster-buster/index.html 获得参与 调节基因转录的序列簇信息 (3) SNP信息分析方法 分析方法:Cygwin analysis program。 该程序通过对基因序列的生物信息进行综合分析, 寻找可能具有各种功能的多态性位点,为遗传学、 分子生物学、进化和系统发育学的研究提供参考数 据或功能信息,对复杂性疾病易感基因的研究工作 很有帮助。 生物信息学分析结果例证 中国汉族群体脂蛋白酯酶基因(LPL)SNP信息