presentation
advertisement

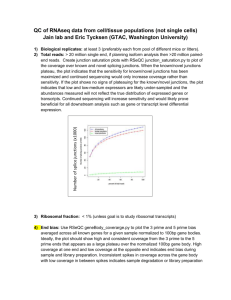
First of all: “Darnit Jim, I’m a doctor not a bioinformatician!” Researcher interested in gene expression • I have obtained raw RNAseq files (FASTQ) for a set of cell lines. How can I process this data and examine my gene(s) of interest? – Ask a bioinformatician – Do it yourself using TraIT tools: run available NGS workflow in Galaxy First time experience of Galaxy Looks like RNA expression analysis… But, I have something called a FASTQ file I don’t know about this format, where do I get such a reference? Looks like RNA expression analysis… And many more options… How do I know that the settings here are correct for my type of data? Instead of a BAM file I have a FASTQ file. How do I process this? Solution: readily available workflow And other pipelines in progress Gene expression: input parameters Ideally metadata on these parameters was provided by original data owners and/or can be traced back (own data known; from other person trace back) Trial run • For 4 colorectal cancer cell lines the FASTQ files were provided. Data owner could provide: • platform • adapter sequences • library type • Wanted to compare these to the processed RNAseq data of prostate cell lines (same experimental platform was used). • Ran workflow and obtained readcounts/measure of expression for the new cell lines. Comparison: colon and prostate Possible for non/little-informed user to run Galaxy workflow and obtain results in a format that can be used in downstream analysis. Further analysis… • Usually, comparison is tumour sample vs normal sample. – EdgeR is available to perform this comparison. • Comparison of expression between groups is possible (e.g. colorectal cell lines vs prostate cell lines), however, when I have only cell lines: – how to solve the question: “does my gene of interest show altered expression in a particular sample compared to a reference sample?” Issues • When not in possession of normal/reference in the dataset (T only, cell lines), how to determine altered expression of a gene of interest? – Use a general normal reference that needs to be provided for comparison? (standard cut-off for increased or decreased expression) < xx reads = decreased exp, > xxx reads = increased exp? – Calculate a median expression for all genes of the platform and then compare expression of one gene to median expression of all genes (significant outliers?) – Distiguish expression of a gene in diploid vs aneuploid cells trouble, in most cases no ploidy status known Issues • When investigating data in the data-integration platform, query for the gene AURKA will give certain results. • If one study had T/N and the other only T – and different manners for determining altered expression were applied – can this data be compared? – Pro: it’s processed and called data you’re comparing in this platform, trust the called data – Con: I don’t think it’s fair to compare differently called data – if comparing such datasets, start from the beginning and treat in the same manner convert the data of the T/N analysed data to T-only or cell line only analysed