Chronic Myeloid Leukemia
Amer Rassam, M.D.
Learning Objectives
Myeloproliferative disorders (MPDs)
Molecular genetics of chronic myeloid
leukemia
Clinical manifestations and diagnosis of
chronic myeloid leukemia
Overview of the treatment of chronic myeloid
leukemia
Initial treatment of chronic myeloid leukemia in
chronic phase
Learning Objectives
Explain how to define and identify a relapse
Treatment of CML in chronic phase after
failure of initial therapy
Clinical use of tyrosine kinase inhibitors for
chronic myeloid leukemia
Treatment of CML in accelerated phase and
blast crisis
Prognosis
Myeloproliferative Disorders
Chronic Myeloid Leukemia (CML)
Polycythemia Vera (PCV)
Essential Thrombocythemia (ET)
Primary Myelofibrosis (PMF)
Myeloproliferative Disorders
Clonal disorders of hematopoiesis that arise in
hematopoietic stem or early progenitor cell.
Characterized by the dysregulated production of
particular lineage of mature myeloid cells with fairly
normal maturation.
Exhibit a variable tendency to progress to acute
leukemia
Share abnormalities of hemostasis and thrombosis
Overlap between the clinical features
Introduction
CML is a clonal myeloproliferative neoplasm
Dysregulated production and uncontrolled proliferation
of mature and maturing granulocyte with fairly normal
differentiation
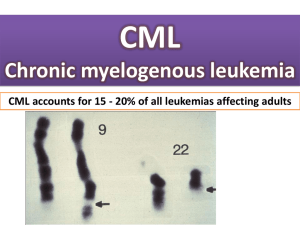
Fusion of 2 genes: BCR (or chromosome 22) and ABL1
(on chromosome 9), resulting in BCR-ABL1 fusion gene
Final result: Abnormal chromosome 22 called
Philadelphia (Ph) chromosome
Final product: BCR-ABL1 fusion protein, a dysregulated
tyrosine kinase
Introduction
Uncontrolled production of mature and maturing
granulocytes
Predominantly neutrophils, but also basophils and
eosinophils
Triphasic or biphasic clinical course
Chronic phase, accelerated phase, blast crisis
Phases of CML (before Imatinib)
Advanced phases
Chronic phase
Median duration
5–6 years
Accelerated
phase
Blast crisis
Median duration
6–9 months
Median survival
3–6 months
Epidemiology
Annual incidence: 1 to 2 cases per 100,000
15% – 20% of all adult leukemias
Incidence increases significantly with age
– Median age: ~ 55 years
– Prevalence increasing due to current therapy
– Most patients present in CP, 85%
• Majority of CML-related deaths due to progression to AP/BC
– 50% of CML patients are asymptomatic at diagnosis
Risk factors
– Exposure to ionizing radiation, the only known
Molecular Genetics of CML
The Philadelphia chromosome was originally detected by
workers in Philadelphia.
The first genetic abnormality to be associated with a
human cancer.
The result of a balanced translocation between
chromosomes 9 and 22.
Derivative chromosome 22 is significantly smaller
Ph chromosome is present in hematopoietic cells from
patients with CML.
Therefore, the Ph chromosome is acquired and NOT
inherited through the germline.
Molecular Genetics of CML
The development of chronic phase CML appears to be a
direct result of the BCR-ABL1 activity, which promotes
its development by allowing:
I.
Uncontrolled proliferation of transformed cells
II.
Discordant maturation
III.
Escape from apoptosis
IV.
Altered interaction with the cellular Matrix
The progression of CML from chronic phase to accelerated face or
blast crisis is a complex, multistep process (may be related to GMP).
Also, it appears to involve the constitutive expression of the BCRABL1 tyrosine kinase.
Molecular Genetics of CML
{
q11
BCR
BCR
ABL
Ph
22
{
q34
ABL
9
ABL
BCR
9q+
Ph chromosome and bcr-abl gene
Chromosome 9
Chromosome 22
9 q+
9
cbcr
1
2-11
c-abl
Ph (or 22q-)
22
bcr
bcr-abl
2-11
p210Bcr-Abl
2-11
p190Bcr-Abl
Exons
abl
FUSION
PROTEIN
WITH
TYROSINE
KINASE
ACTIVITY
t(9;22) translocation
Introns
CML Breakpoints
ALL Breakpoints
bcr-abl gene structure
Philadelphia chromosome
t(9;22)(q34;q11)
22q- = Philadelphia chromosome
Clinical Manifestations
Asymptomatic in 20-50% of patients
Fatigue 34%, weight loss 20%, excessive sweating
15%, abdominal fullness 15%, bleeding episodes
21% (platelet dysfunction).
Abdominal pain in the LUQ (enlarged spleen)
Tenderness over the lower sternum.
Acute gouty arthritis
Findings: Splenomegaly, anemia, WBC > 100,000,
platelet count > 600,000
Peripheral Blood Pathology
Leukocytosis (median of 100,000)
Differentiation shows virtually all cells of neutrophilic
series
Blasts < 2%
Myelocytes more than metamyelocytes (a classic finding
in CML)
Neutrophils cytochemistry is abnormal – low LAP score
Basophilia in 90% of cases
Thrombocytosis. If low platelets – consider an other
CML Peripheral Blood Smear
CML Peripheral Blood Smear
Bone Marrow Pathology
Granulocytic maturation pattern same as in the
peripheral blood
Increased reticulin fibrosis and vascularity
Erythroid islands are reduced in number and size
Dwarf megakaryocytes
Pseudo-Gaucher’s cells and Sea Blue histiocytes
(markers of increased cell turnover)
Iron-laden macrophages are reduced or absent
Pseudo-Gaucher cells
Pseudo-Gaucher cells
Sea Blue Histiocyctes
CML – Bone Marrow
Diagnosis of CML
Typical findings in the blood and bone marrow
Requires the detection of the Ph chromosomal or its
product, the BCR-ABL1 fusion mRNA and the BCR-ABL1
protein.
Conventional cytogenetic analysis (karyotyping) – The first
method
Florence and in situ hybridization (FISH) analysis
RT-PCR (The BEST)
Southern blot techniques – rarely used
Western Blotting – low sensitivity and labor intensive
BCR-ABL (FISH)
RT-PCR for BCR-ABL
Qualitative RT-PCR allow for
the diagnosis of CML
Quantitative RT-PCR is used
to quantify the amount of
disease
Target
sequence
1 Denaturation:
Heat briefly to
separate DNA
strands
2 Annealing: Cool
Cycle 1
yields 2
molecules
Allows for the identification
of cryptic BCR-ABL
translocations
Does not require a bone
marrow aspirate for optimal
results
to allow primers
to form hydrogen
bond with ends
of target
sequence
Primers
3 Extension: DNA
polymerase adds
nucleotides to the
3” end of each
primer
Cycle 2
yields 4
molecules
Cycle 3 yields
8 molecules;
2 molecules
(in white
boxes)
match target
sequence
New
nucleotides
Most CML patients are diagnosed
in the chronic phase
Chronic phase
Blastic phase
Differential Diagnosis
Leukemoid reaction
Juvenile myelomonocytic leukemia (JMML)
Chronic myelomonocytic leukemia (CMML)
Atypical CML
Chronic eosinophilic leukemia
Chronic neutrophilic leukemia
Other myeloproliferative neoplasms
Other Ph chromosome positive malignancies
Accelerated Phase CML
10-19% blasts in the peripheral blood or bone
marrow
Peripheral blood basophils ≥20%
Platelets < 100,000/microL, unrelated to therapy
Platelets > 1,000,000/microL, unresponsive to
therapy
Progressive splenomegaly and increasing WBC,
unresponsive to therapy
Cytogenic evolution
Blastic Phase CML
Blast crisis is generally refractory to
treatment, occurs approximately 3-5 years
after the diagnosis of CML and 18 months
after the onset of accelerated face
Blasts in the peripheral blood ≥20% or in the bone
marrow ≥30%
Large foci or clusters of blasts on the bone marrow
biopsy
Presence of extramedullary blastic infiltrate (e.g.,
myeloid sarcoma, also known as granulocytic
sarcoma or chloroma)
Blast Phase CML – Bone Marrow
Blast Phase CML – Bone Marrow
Clinical Debate
What is the optimal frontline
therapy for CML?
Principles of CML treatment
Relieve symptoms of hyperleukocytosis,
splenomegaly and thrombocytosis.
Hydration
Chemotherapy (Busulfan, hydroxyurea)
Control and prolonging the chronic phase (noncurative)
Tyrosine kinase inhibitors
Alpha-interferon + chemotherapy
Chemotherapy (hydroxyurea)
Treatment Options
Treatment decisions for patients with CML
are complex, due to the variety of available
options, many of which are conflicting.
Potential cure with allogeneic
hematopoietic stem cell transplantation
Disease control without cure using
tyrosine kinase inhibitors (TKIs)
Palliative therapy with cytotoxic agents
Factors influencing choice of
therapy
Phase of CML
Availability of a donor for allogeneic stem cell
transplant
Patient age
Presence of medical co-morbidities
Response to treatment with TKIs
IRIS Study Design: Imatinib Mesylate
Versus IFN- + ara-C
1106 patients enrolled from June 2000 to January 2001
Imatinib Mesylate
S
R
IF:
Loss of MCR or CHR
Increasing WBC count
Intolerance of treatment
Failure to achieve MCR at 12 months*
Failure to achieve CHR at 12 months*
Request to discontinue IFN-*
Crossover
IFN- + ara-C
S = screening.
R = randomization.
Progression
Increasing WBC count
Loss of MCR or CHR
Accelerated phase or blast crisis
Death
Hematologic Responses
100
96%
Imatinib mesylate
90
% Responding
80
70
60
67%
IFN- + ara-C
50
40
30
20
10
0
0
3
6
9
12
15
Months Since Randomization
18
21
Cytogenic Responses
100
90
83%
Imatinib mesylate
% Responding
80
70
60
50
40
30
20%
IFN- + ara-C
20
10
0
0
3
6
9
12
15
Months Since Randomization
18
21
Overall Survival on First-Line Imatinib
(IRIS Study)
Resistance to Imatinib occurs predominantly
during advanced phase CML
Patients in advanced
phase often relapse with
the development of
chemotherapy resistance
Some patients in blast
crisis CML respond to
Imatinib but then tends
to relapse
Chronic
Phase
Blast
Crisis
Relapse
Hematopoietic
differentiation
Advanced stage cancers
are characterized by
multiple genetic changes
Bone marrow to
peripheral blood
Ph-negative
Ph+ blasts
Ph+
Ph+ Imatinib mesylateresistant blasts
Initial Treatment
Tyrosine kinase inhibitors are for first-line
therapy in chronic phase CML
Imatinib (Gleevec)
Dasatinib (Sprycel)
Nilotinib (Tasigna)
1.
2.
All 3 agents are considered to be (category 1) based on the NCCN
guidelines and recommendations.
Second-generation TKIs (dasatinib or nilotinib) produce faster and
deeper response than imatinib
Treatment of CML after failure of
initial therapy
No randomized trials have directly compared the
efficacy of second-generation TKIs in patients
with chronic phase CML who experience failure
of an initial TKIs
A trial of another TKI.
Dasatinib preferred in patients with pancreatitis,
elevated bilirubin or hyperglycemia
Dasatinib crosses the blood brain barrier and would
therefore be preferred in patients with CNS involvement
Nilotinib might be chosen for patients with a history of
pleural or pericardial effusion or disease
Dasatinib and Nilotinib can result in QT prolongation
Other Options
Bosutinib – toxicity is a limiting factor
Ponatinib – toxicity is a limiting factor
Increase the dose of Imatinib
Omacetaxine mepesuccinate – SQ Injection
Approved by the FDA for patients resistant or
intolerant to 2 or more TKIs
Hematopoietic cell transplant – the only cure
Clinical trials
Other Options
Patients who are ineligible for HCT but have
either a contraindication to a second-generation
TKI or have failed to respond to treatment with
available TKI
Interferon alfa plus cytarabine
Hydroxyurea
Busulfan
Response Criteria
Hematologic response
Cytogenic response
Molecular response
Resistance to treatment
Primary resistance – patient fails to
achieve a desired response to initial
treatment
Secondary resistance – patient with an
initial response to a TKI ultimately
relapses
Loss of Response
Patients should be re-evaluated with a bone
marrow biopsy with cytogenetics, and BCR-ABL
kinase mutation analysis
T315I mutation
Resistant to all TKIs, except Ponatinib
Patient should be evaluated for SCT
Y253H, E255k/V and F359V/C/I mutations
Resistant to Imatinib and Nilotinib but sensitive to Dasatinib
F317L/V/I/C, V299L and T315A mutations
Sensitive to Nilotinib but with intermediate sensitivity to
Imatinib and Dasatinib
Mechanisms of action TKIs
They block the initiation of bcr-abl pathway
Many TKIs also affect other signaling pathways
Dasatinib and Bosutinib inhibit both Bcr-Abl and Src
kinases.
Nilotinib inhibits Bcr-Abl, c-kit and platelet derived
growth factor receptor (PDGFR)
These differences in targeted pathways may be
responsible for their varied clinical effects in tumors
Mechanisms of Action, Imatinib
Competitively inhibits the inactive configuration of
the Bcr-Abl protein tyrosine kinase
Blocking the ATP binding site and thereby
preventing a conformational switch to the active
form
Inhibits cellular proliferation and tumor formation
Produces 95% decrease in CML colony growth
Inhibits platelet-derived growth factor and c-kit
GLEEVEC (Imatinib)
GLEEVEC (Imatinib)
Molecular consequence
of the t(9;22) is the fusion
protein BCR–ABL, which
has increased in tyrosine
kinase activity
BCR-ABL protein
transform hematopoietic
cells so that their growth
and survival become
independent of cytokines
It protects hematopoietic
cells from programmed
cell death (apoptosis)
TASIGNA (Nilotinib)
Drug Interaction with TKIs
They are metabolized by the CYP3A4 system – can
inhibit other cytochrome P450 pathways
Therefore, they compete with Coumadin
Low TKIs levels – St. John’s wort, rifampin,
carbamazepine, phenobarbital and phenytoin
High TKIs levels – diltiazem, verapamil, itraconazole,
ketoconazole, clarithromycin, erythromycin and
grapefruit juice
Side Effects of TKIs
Imatinib - Bone marrow suppression; fluid retention/edema;
gastrointestinal effects; heart failure; hepatotoxicity
Dasatinib - Bone marrow suppression; pleural/pericardial
effusions; pulmonary arterial hypertension; QT prolongation;
aspirin like effect
Bosutinib - Bone marrow suppression; fluid retention/edema;
gastrointestinal effects
Side Effects of TKIs
Nilotinib - Bone marrow suppression;
atherosclerosis-related events; electrolyte
imbalance; hepatotoxicity
Black box: QT prolongation (screening required)
Ponatinib - Bone marrow suppression; fluid
retention/edema; gastrointestinal effects; heart
failure; hypertension; pancreatitis; aspirin-like effect
Black box: Arterial thrombosis; hepatic toxicity
Pregnancy and TKIs
All TKIs could be teratogenic during pregnancy
Women are advised not to become pregnant
while on TKIs (any TKI)
Best effective contraception is the barrier
Woman taking TKIs are advised to avoid to
breast-feeding
Prognosis
Improved dramatically since the incorporation of
tyrosine kinase inhibitors into the initial treatment
SEER database. 5138 patient’s, year 2000 and 2005
15-44 years – OS 72 versus 86%
45-64 years – OS 68 versus 76%
65-74 years – OS 38 versus 51%
75-84 years – OS 19 versus 36%
Stage of disease at the time of diagnosis is the
strongest single predictor of outcome.
www.tallahasseecancer.com
www.tallahasseecancer.com