Myeloproliferative disorders
advertisement

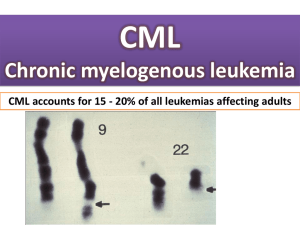
Myeloproliferative disorders (MPD) are chronic diseases caused by clonal proliferation of bone marrow stem cells leading to excess production of one or more haemopoietic lineage. The clinical syndromes include polycythaemia rubra vera (red cells), essential thrombocythaemia (platelets), chronic myeloid leukaemia (white cells) and myelofibrosis in which there is a reactive fibrosis of the marrow and extramedullary haemopoiesis in the liver and spleen. Intermediate forms may occur and the diseases may all transform into acute myeloid leukaemia. Chronic myeloid leukaemia This is a clonal myeloproliferative disorder characterized by an increase in neutrophils and their precursors in the peripheral blood with increased cellularity of the marrow as a result of an excess of granulocyte precursors. Without treatment, CML progresses from an initial chronic phase (CP) characterized by marrow hyperplasia and increased numbers of circulating differentiated myeloid cells followed by advanced phases of disease (accelerated phase [AP] and blast crisis [BC]) marked by a block in differentiation, an accumulation of blasts, and a depletion of normal hematopoietic cells, especially white blood cells and platelets. CML was the first malignant disease found to be consistently associated with a specific cytogenetic abnormality, the Philadelphia chromosome (Ph), resulting in the formation of the BCR–ABL fusion oncogene. Study of BCR–ABL has led to sensitive methods to detect residual disease and predict outcome and to “targeted” therapy aimed at inhibiting abnormal tyrosine kinase activity resulting from the BCR–ABL fusion oncogene. In addition, CML was one of the first diseases demonstrated to be curable by hematopoietic cell transplantation (HCT). Aetiology Aetiology is unknown. Radiation may play a role in some cases, because persons exposed to high-dose irradiation, including survivors of the atomic bomb, have a significantly increased risk of leukemia, and high-dose irradiation of myeloid cell lines in vitro induces the expression of BCR–ABL transcripts indistinguishable from those that characterize CML. Pathophysiology Chronic myelogenous leukemia was recognized as a distinct entity, associated with massive splenomegaly and leukocytosis without other explanations, in the mid1800s. The modern history of CML was initiated by Nowell and Hungerford in 1960. They used newly developed techniques to detect a small chromosome in metaphase preparations of marrow cells from CML patients. This abnormal chromosome was the first consistent chromosomal abnormality in human malignancies and was termed the Philadelphia chromosome after the city of its discovery. Rowley showed that the Philadelphia chromosome resulted from a translocation between chromosomes 9 and 22 [t(9; 22)(q34;q11)]. The genes involved in this translocation were cloned in the 1980s, and the t(9 : 22) translocation was shown to result from the fusion of the BCR (breakpoint cluster region) gene on chromosome 22 to the ABL (Abelson leukemia virus) gene on chromosome 9, with formation of the BCR–ABL fusion oncogene. This fusion gene encodes a 210-kDa protein with greatly increased tyrosine kinase activity, which is now believed to be the principal cause of the chronic phase of CML. The disease is of stem cell origin as the Ph chromosome is present in erythroid, granulocytic, megakaryocytic and T-lymphoid precursors. Chronic myelogenous leukemia is the most common of the myeloproliferative diseases and represents 15% to 20% of all new leukemia cases. The annual incidence of CML is 1 to 1.5 cases per 100,000 population per year. The median age at diagnosis is 67 years and the incidence sharply rises with age. The disease occurs slightly more often in men than in women. Chronic myelogenous leukemia may occur in children but only approximately 10% of cases occur in subjects between 5 and 20 years of age, and represent only 3% of all childhood leukemias. The disease usually transforms from a relatively stable chronic phase to an acute leukaemia phase (blast transformation). Phases Chronic 2. Accelerated phase 3. Blast crisis 1. Most (>90%) CML patients present in chronic phase (CP). CML is often diagnosed incidentally during routine examination or examination for another illness. Symptoms Presenting symptoms include weight loss, night sweats, itching, left hypochondrial pain, gout. Priapism, visual disturbance and headaches caused by hyperviscosity (WBC >250 Ґ 109/L) are less frequent. Splenomegaly, often massive, occurs in over 90% of cases. Laboratory Findings Raised white cell count (often 50 Ґ 109/L or more), mainly neutrophils and myelocytes (granulocytes at all stages of differentiation in peripheral blood cells. Circulating granulocytes are usually normal in appearance, but with low leucocyte alkaline phosphatase score). Although the proportion of eosinophils is usually not increased, the absolute eosinophil count is usually increased. The absolute basophil count is almost always increased in CML. The proportion of basophils is usually less than 15% in chronic-phase patients, but may rarely be higher. The platelet count is elevated in 50% of patients at the time of diagnosis. Thrombocytopenia is rare at diagnosis and usually is a sign of progression toward accelerated phase. Raised serum uric acid. Bone marrow is hypercellular. The granulocytic/erythroid ratio is increased to 10 : 1 to 30 : 1, with increased granulopoiesis and reduced erythropoiesis. Presence of more than 10% blasts indicates transformation to accelerated phase. Cytogenetic analysis of bone marrow cells shows the Philadelphia chromosome in >95% of metaphases. The BCR-ABL fusion gene is detectable by FISH or polymerase chain reaction (PCR) assays. Fluorescent in situ hybridization (FISH). Accelerated Phase General, accelerated phase is characterized by symptoms of fever, night sweats, weight loss and bone pain, difficulty in controlling counts using conventional therapy, increased numbers of blasts and early myeloid cells in marrow and peripheral blood, and evidence of karyotypic evolution Blast Crisis The blast phase of CML resembles acute leukemia. Blast crisis is defined as having more than 20% blasts in the bone marrow or peripheral blood, the presence of large aggregates and clusters of blasts in the bone marrow biopsy, or the development of extramedullary blastic infiltrates. Blasts may be of myeloid or lymphoid lineage. Course and progress Patients are typically well during the ‘chronic phase’. The main cause of death is transformation into acute leukaemia, which may occur at any stage, even at presentation. Median survival is currently about 4 years. Staging to predict prognosis uses age, spleen size, blood blast cell and platelet counts. There may be an accelerated phase of variable duration in which anaemia, thrombocytopenia, splenic enlargement and marrow fibrosis occur. Transformation is usually accompanied by additional morphological and chromosome abnormalities Hematologic Response A complete hematologic remission is defined as the achievement of normal WBC and platelet counts and normal differential, and disappearance of all symptoms and signs of CML.[ A partial hematologic response is defined as a decrease in the WBC count to less than 50% of the pretreatment level, or the normalization of the WBC count accompanied by persistent splenomegaly or immature cells in the peripheral blood. Cytogenetic Response Complete: Ph-negative = 0% Major: Ph-positive = 1% to 35% Minor: Ph-positive = 36% to 65% Minimal: Ph-positive = 66% to 95% None: Ph-positive < 95% Molecular Response Complete: BCR–ABL transcripts nonquantifiable and nondetectable Major: BCR–ABL transcripts ≤0.10% Treatment. Chronic phase Busulfan (BU) chemotherapy for CML was introduced in the 1950s. Busulfan was administered in doses of 4 to 6 mg/day and then held when the WBC count fell to 30 × 109/L. The drug effect could persist for weeks, and the counts could fall further after therapy was discontinued. Busulfan therapy was associated with serious adverse effects, including prolonged aplasia, pulmonary fibrosis, and a syndrome simulating adrenal insufficiency. Treatment. Chronic phase Treatment with hydroxyurea (HU) was started as an alternative to busulfan. Hydroxyurea therapy is usually initiated at doses of 1 to 6 g/day in an attempt to lower counts. Hydroxyurea administered at doses of 1 to 2 g/day is then used to maintain blood counts in the normal range. Hydroxyurea is less toxic than busulfan. Its major adverse effect is reversible marrow suppression. Because neither drug results in significant selective suppression of the Ph-positive clone, the aim of therapy with these agents is to control disease and symptoms. Hydroxyurea is now commonly used to achieve control of counts simultaneous with or prior to initiation of treatment with imatinib or other disease-specific therapies. Treatment. Chronic phase α-interferon (IFN) may also control the white cell count and may delay the onset of acute transformation, prolonging overall survival by 1–2 years. The best responders to IFN become Ph-negative, but usually remain BCR-ABL-positive, and have the best prognosis. The potential mechanisms by which IFN works in CML are not understood, but may include inhibition of increased proliferation, correction of the adhesion defect of the malignant progenitor in CML, or stimulating an immune response to CML. Rates for complete and partial cytogenetic remissions range from 0% to 38%. Treatment. Chronic phase Because the tyrosine kinase activity of BCR–ABL plays a critical role in cellular transformation, it is an attractive target for inhibition. Imatinib (Glivec). This is a specific inhibitor of the tyrosine kinase encoded by BCR-ABL. It controls the blood count and causes the marrow to become Ph negative in a high proportion of cases, though nearly all remain positive for the BCR-ABL fusion messenger RNA when tested by PCR. The chronic phase is prolonged and the rate of acute transformation is reduced. Side effects include nausea, skin rashes and muscle pains. Imatinib in combination with other drugs is also valuable in the therapy of Ph+ALL and blast transformation of CML. Glivek At a chronic stage 80-91% have a complete hematologic remission, 49-61% have major cytogenetic response and 3036% have complete elimination of Ph+ cells At accelerated phase: 63-69% have a complete hematologic remission, 20-24% have major cytogenetic response and 1417% have complete elimination of Ph+ cells While prolongating glivek treatment (9-12 months) the rate of major cytogenetic response rises to до 74-81%. Even in the blast crisis the Glivec treatment gives positive results: 26-29% have a complete hematologic remission, 4961% have major cytogenetic response and 6-7% have complete elimination of Ph+ cells The duration of Glivec treatment 3 months – complete hematologic remission 6 months– major cytogenetic response 12 months– complete cytogenetic response 18 months– major molecular response An alternative to Glivek Tasigma (nilonitib) – the second-line drug for patients at a chronic phase and at an accelerated phase. Is used in case of resistancy to Glivec. 50% of patients have major cytogenetic response. Treatment. Chronic phase Allogeneic stem cell transplantation (SCT) before the age of 50 from an HLA matching sibling offers a 70% chance of cure in the chronic phase but 30% or less once acceleration has occurred. HLA-matched unrelated donor (MUD) SCT is less successful in curing the disease because of higher morbidity and mortality. Transfusion of donor lymphocytes may be valuable in eliminating BCRABLpositive cells in cases of relapse post-SCT. Treatment. Acute phase Therapy as for acute leukaemia, AML or ALL with the addition of imatinib may be given, but the prognosis is poor. Myelofibrosis Myelofibrosis (myelosclerosis, agnogenic myeloid metaplasia) is characterized by splenomegaly, extramedullary haemopoiesis, a leucoerythroblastic blood picture and replacement of bone marrow by collagen fibrosis. Myelofibrosis is a clonal disease as well. In PMF, there is a profound hyperplasia of morphologically abnormal megakaryocytes and clonal populations of monocytes that may be responsible for the marrow fibrosis due to the local release of fibrogenic growth factors Aetiology and pathophysiology The primary defect is within the haemopoietic stem cell; Chromosome abnormalities are common, but they are different Fibrosis results from a reactive non-neoplastic proliferation of marrow stromal cells. Very important feature – the ability of malignant cells for extramedullar spread, infiltration with malignant cells of the spleen, liver, lymph. nodes, others. Epidemiology 0,5-1,5 cases per 100,000 persons per year The average age at diagnosis of PMF is approximately 65 years, and most patients are diagnosed between 50 and 69 years of age. Clinical features The first stage (onset): panmyelosis is common; Symptoms due to erythrocytosis, thrombocytosis (Thrombotic episodes ). Thrombosis may be venous (cerebral venous sinus thrombosis, splanchnic vein thrombosis, deep vein thrombosis, pulmonary thromboembolism) or arterial (stroke, transient ischemic attacks, retinal artery occlusion, myocardial infarction, angina pectoris, and peripheral arterial disease). The cellular phase of PMF with thrombocytosis and presence of cardiovascular risk factors such as hypertension, smoking, hypercholesterolemia, and diabetes are the independent predictors of thrombosis. Hyperviscosity may lead to headaches and visual disturbance. Spleen is not enlarged very much. This stage has much in common with polycythemia vera and is often called as subleucemic myelosis. Clinical features The second, fibrotic stage Splenomegaly is very massive. Spleen often takes half of the stomach. Patients may merely complain of a dull, heavy sensation in the left upper quadrant. Pain of extreme severity, simulating an acute abdominal emergency, is produced by splenic infarction. Anemia (myelofibrosis, hypersplenism) Haemorrhage. Bleeding may be trivial, as manifested by petechiae and ecchymoses, or it may be life-threatening as a result of uncontrollable esophageal bleeding. It may result from thrombocytopenia or poor platelet function The terminal stage: Polyorganic complications. The reason of death may be acute esophageal bleeding (portal hypertension) Transformation into acute leycosis in some patients Laboratory features Normochromic normocytic anaemia. Leucocytosis and thrombocytosis with circulating megakaryocyte fragments occur early; leucopenia and thrombocytopenia occur later. Blood film: red cell poikilocytosis with teardrop forms and circulating red cell and white cell precursors (leucoerythroblastic picture) Serum LDH is raised. Liver function tests are often abnormal because of extramedullary haemopoiesis. NAP score is usually raised. Bone marrow aspiration is usually unsuccessful (‘dry tap’); the trephine biopsy shows increased cellularity, increased megakaryocytes and fibrosis Treatment Chemotherapy (e.g. hydroxyurea) for patients with hypermetabolism and myeloproliferation. Thalidomide improves marrow function and reduces spleen size in about a third of cases. Supportive therapy with red cell transfusions, folic acid and occasionally platelet transfusions. Iron chelation may be needed. Allopurinol to prevent hyperuricaemia and gout. Splenectomy or splenic irradiation to reduce symptoms from splenomegaly, anaemia or thrombocytopenia (selected patients only). Allogeneic bone marrow transplantation has cured a few younger patients (<50 years). Prognosis Median survival is about 5 years; acute leukaemia occurs in about 20%. POLYCYTHEMIA VERA PV is a clonal, chronic, progressive myeloproliferative disorder (MPD) often of insidious onset, characterized by an absolute increase in red cell mass and also usually by leukocytosis, thrombocytosis, and splenomegaly. PV leads to excessive proliferation of erythroid, myeloid, and megakaryocytic elements within the bone marrow. Epidemiology 0,6-1,6 cases per 100,000 persons per year The average age at diagnosis of PMF is approximately 60 years. May occur in young people Clinical features Raised RCM causes a ruddy complexion and conjunctival suffusion; hyperviscosity may lead to headaches and visual disturbance. Thrombosis (e.g. deep vein thrombosis (DVT), Budd– Chiari syndrome, which results from hepatic venous or inferior vena caval thrombosis and obstruction) is also caused by hyperviscosity and increased platelets. Haemorrhage, especially gastrointestinal, may occur. Excess histamine secretion from basophils leads to increased gastric acid and peptic ulcer is frequent. Polycythaemia rubra vera: patient with plethora. Arterial thrombotic events account for two-thirds of such events, with venous thrombotic events representing the remainder. Ischemic stroke, myocardial infarction, and transient ischemic attacks are the most common arterial thrombotic events. Patients may also present with deep venous thrombosis in the lower extremities, pulmonary embolism, or peripheral vascular occlusions Differential diagnosis Secondary or reactive polycythaemia may occur in conditions where arterial oxygen saturation is reduced, leading to a physiological rise in EPO, or when EPO levels are inappropriately raised (e.g. caused by secretion of EPO by a renal neoplasm). Relative polycythemia 1. Decreased plasma volume—reduced fuid intake, marked loss of body fuids (diaphoresis, vomiting, diarrhea, “third-spacing”) 2. Overfilling of blood in collection vacuum tubes AbsolutepPolycythemia Secondary polycythemia : Pulmonary disease Hypoventilation syndromes—sleep apnea, Pickwickian syndrome Smokers' polycythemia, carbon monoxide intoxication due to industrial exposure Postrenal transplantation erythrocytosis Tumors—renal cell carcinoma, Wilms tumor, hepatic carcinoma, uterine leiomyomata, virilizing ovarian tumors, vascular cerebellar tumors Miscellaneous renal and hepatic disorders—solitary renal cysts, polycystic kidney disease, renal artery stenosis hydronephrosis, viral hepatitis Endocrine disorders—Cushing's syndrome, primary aldosteronism Androgen use Erythropoietin use The following additional tests are occasionally required: Chest X-ray; arterial blood gas analysis to exclude lung disease. Haemoglobin oxygen dissociation curve to identify a variant haemoglobin with increased oxygen affinity. Serum EPO assay. Treatment Thrombosis is the main cause of morbidity and mortality and its incidence can be reduced by maintaining the PCV below 0.45 and platelets below 600 Ґ 109/L. Aspirin (75 mg daily) is often used to inhibit platelet function. Regular venesection is used initially to lower the PCV. Busulfan may be given orally. It has a more prolonged action than hydroxyurea and more side-effects and is now more rarely used. Prognosis Median survival is about 16 years. Up to 30% of patients develop myelofibrosis . Acute myeloid leukaemia occurs in up to 5% of patients