Lecture Slides - School of Chemical Sciences
advertisement

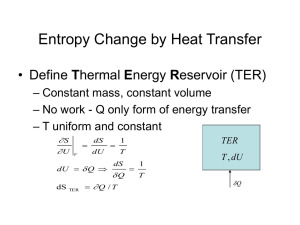
Chemistry 444 Chemical Thermodynamics and Statistical Mechanics Fall 2006 – MWF 10:00-10:50 – 217 Noyes Lab Instructor: Prof. Nancy Makri Teaching Assistant: Adam Knapp Office: A442 CLSL E-mail: nancy@makri.scs.uiuc.edu Office Hours: Fridays 1:30-2:30 (or by appointment) Office: A430 CLSL E-mail: aknapp2@uiuc.edu Office Hours: Mondays 1:30-2:30 http://www.scs.uiuc.edu/~makri/444-web-page/chem-444.html Why Thermodynamics? The macroscopic description of a system of ~1023 particles may involve only a few variables! “Simple systems”: Macroscopically homogeneous, isotropic, uncharged, large enough that surface effects can be neglected, not acted upon by electric, magnetic, or gravitational fields. Only those few particular combinations of atomic coordinates that are essentially time-independent are macroscopically observable. Such quantities are the energy, momentum, angular momentum, etc. There are “thermodynamic” variables in addition to the standard “mechanical” variables. Thermodynamic Equilibrium In all systems there is a tendency to evolve toward states whose properties are determined by intrinsic factors and not by previously applied external influences. Such simple states are, by definition, time-independent. They are called equilibrium states. Thermodynamics describes these simple static equilibrium states. Postulate: There exist particular states (called equilibrium states) of simple systems that, macroscopically, are characterized completely by the internal energy U, the volume V, and the mole numbers N1, …, Nr of the chemical components. The central problem of thermodynamics is the determination of the equilibrium state that is eventually attained after the removal of internal constraints in a closed, composite system. Laws of Thermodynamics What is Statistical Mechanics? Link macroscopic behavior to atomic/molecular properties Calculate thermodynamic properties from “first principles” (Uses results for energy levels etc. obtained from quantum mechanical calculations.) The Course Discovery of fundamental physical laws and concepts An exercise in logic (description of intricate phenomena from first principles) An explanation of macroscopic concepts from our everyday experience as they arise from the simple quantum mechanics of atoms and molecules. …not collection of facts and equations!!! The Course Prerequisites • • Tools from elementary calculus Basic quantum mechanical results Resources • • • • “Physical Chemistry: A Molecular Approach”, by D. A. McQuarrie and J. D. Simon, University Science Books 1997 Lectures (principles, procedures, interpretation, tricks, insight) Homework problems and solutions Course web site (links to notes, course planner) Course Planner o o o o o Organized in units. Material covered in lectures. What to focus on or review. What to study from the book. Homework assignments. Questions for further thinking. http://www.scs.uiuc.edu/~makri/444-web-page/chem-444.html/444-course-planner.html Grading Policy Homework 30% (Generally, weekly assignment) Hour Exam #1 15% (September 29th) Hour Exam #2 15% (November 3rd) Final Exam (December 14th) 40% Please turn in homework on time! May discuss, but do not copy solutions from any source! 10% penalty for late homework. No credit after solutions have been posted, except in serious situations. Math Review • • • • Partial derivatives Ordinary integrals Taylor series Differential forms Differential of a Function of One Variable f df df dx x df dx dx Differential of a Function of Two Variables f f ( x0 d x, y0 ) f ( x0 , y0 ) d x x y f f ( x0 d x, y0 d y ) f ( x0 d x, y0 ) d y y x f f f f ( x0 , y0 ) d x d y x y y x f (x0, y0) y (x0, y0+dy) (x0, y0) df f ( x0 d x, y0 d y ) f ( x0 , y0 ) (x0+dx, y0+dy) (x0+dx, y0) x f f dx d y x y y x Special Math Tool If z z ( x, y ), then x y z y z x 1 z x y PROPERTIES OF GASES The Ideal Gas Law PV nRT or PV RT , V V /n Extensive vs. intensive properties Units of pressure Units of temperature 1 Pa 1 N m 2 1 atm 1.01325 105 Pa 1 bar = 105 Pa 1 1 torr = atm 760 Triple point of water occurs at 273.16 K (0.01oC) Deviations from Ideal Gas Behavior PV z RT T=300K “compressibility factor” Ideal gas: z = 1 z < 1: attractive intermolecular forces dominate z > 1: repulsive intermolecular forces dominate Van der Waals equation a P 2 V b RT V At fixed P and T, V is the solution of a cubic equation. There may be one or three real-valued solutions. The set of parameters Pc, Vc, Tc for which the number of solutions changes from one to three, is called the critical point. The van der Waals equation has an inflection point at Tc. Isotherms (P vs. V at constant T) P T Tc : 0 V T P 0 : unstable region V T Large V: ideal gas behavior. Only one phase above Tc. Unstable region: liquid+gas coexistence. Critical Point of van der Waals Equation RT 2a P (V b) 2 V 3 V T 2a (V b) 2 0 for V RT 3 (3 or 1 real roots) 2P 2 RT 6a 2 3 4 V T (V b) V 3a (V b)3 0 for V RT 4 Both are satisfied at the critical (inflection) point, so... Vc 3b 8a 27 Rb a Pc 27b 2 Tc The law of corresponding states Eliminate a and b from the van der Waals equation: 2 b 13 Vc , a 27b 2 Pc 3PV c c , 3 1 8 P V R TR 2 R V 3 3 R PR P V T , VR , TR "reduced" variables Pc Vc Tc All gases behave the same way under similar conditions relative to their critical point. (This is approximately true.) Virial Coefficients Z PV B B 1 2V 3V2 RT V V Virial expansion B2V : second virial coefficient, B3V : third virial coefficient,etc. Using partition functions, one can show that B2V (T ) 2 N A 0 e u ( r ) / k BT 1 r 2 dr u (r ): rotationally averaged intermolecular potential Simple Models for Intermolecular Interactions (a) Hard Sphere Model u (r ) , r 0, r 2 B2V (T ) N A 3 3 (b) Square Well Potential , r u (r ) , r 0, r 2 B2V (T ) N A 3 1 ( 3 1) e / kBT 1 3 (c) Hard Sphere Potential with r-6 Attraction , r u (r ) c6 6 , r r B2V (T ) 2 c N A 3 6 3 (high T ) 3 kBT Interpretation of van der Waals Parameters From the van der Waals equation, … B2V (T ) b a RT Comparing to the result of the square well model, 2 b N A 3 , 3 2 a N A2 3 3 1 3 Comparing to the result of the hard sphere model with r-6 attraction, 2 b N A 3 , 3 b a 2 c a N A2 63 3 molecular diameter molecular volume (repulsive interaction) related to strength/range of attractive interaction The Lennard-Jones Model c6 c u (r ) 4 12 12 r6 r Attractive term: dipole-dipole, or dipole-induced dipole, or induced dipoleinduced dipole (London dispersion) interactions. Origin of Intermolecular Forces Hˆ Tˆnucl R i Hˆ el ri , R i Only Coulomb-type terms! ri : electronic coordinates R i : nuclear coordinates The Born-Oppenheimer Approximation: Electrons move much faster than nuclei. Fixing the nuclear positions, Hˆ el ri ,R i n ri ;R i En R i n ri ;R i Adiabatic or electronic or Born-Oppenheimer states Electronic energies; form potential energy surface. Responsible for intra/intermolecular forces. INTRODUCTION TO STATISTICAL MECHANICS The concept of statistical ensembles An ensemble is a collection of a very large number of systems, each of which is a replica of the thermodynamic system of interest. The Canonical Ensemble A collection of a very large number A of systems (of volume V, containing N molecules) in contact with a heat reservoir at temperature T. Each system has an energy that is one of the eigenvalues Ej of the Schrodinger equation. A state of the entire ensemble is specified by specifying the “occupation number” aj of each quantum state. The energy E of the ensemble is E = a E j j j The principle of equal a priori probabilities: Every possible state of the canonical ensemble, i.e., every distribution of occupation numbers (consistent with the constraint on the total energy) is equally probable. How many ways are there of assigning energy eigenvalues to the members of the ensemble? In other words, how many ways are there to place a1 systems in a state with energy E1, a2 systems in a state with energy E2, etc.? Recall binomial distribution: The number of ways A distinguishable objects can be divided into 2 groups containing a1 and a2 =A -a1 objects is A ! W (a1 , a2 ) a1 !a2 ! Multinomial distribution: The number of ways A distinguishable objects can be divided into groups containing a1, a2,… objects is a k ! A ! W a1 , a2 , k a1 !a2 ! ak ! k 3.5 3 25 A 3 A 6 20 2.5 15 W W 2 1.5 10 1 5 0.5 0 0 0 1 2 3 0 1 2 3 4 5 6 a1 a1 1.4 1014 1000 A 12 1.2 10 14 A 50 800 1 1014 600 W W 8 1013 6 10 400 13 4 1013 200 2 10 13 0 0 0 1 2 3 4 5 6 7 8 9 10 11 12 a1 4 14 24 a1 34 44 The Method of the Most Probable Distribution The distribution peaks sharply about its maximum as A increases. To obtain ensemble properties, we replace the weighted average by the most probable distribution. To find the most probable distribution we need to find the maximum of W subject to the constraints of the ensemble. This requires two mathematical tools, Stirling’s approximation and Lagrange’s method of undermined multipliers. Stirling’s Approximation This is an approximation for the logarithm of the factorial of large numbers. The results is easily derived by approximating the sum by an integral. ln N ! N ln N N Lagrange’s Method of Undetermined Multipliers Extremize the function f ( x1 , , xn ) subject to the constraint g ( x1 , , xn ) 0. f d x j 0. j 1 x j n The function has an extremum if d f g d x j 0. j 1 x j n The constraint condition is satisfied at all points, so d g This relation connects the variations of the variables, so only n-1 of them are independent. We introduce a parameter and combine the two relations into f g x j j 1 x j n d x j 0. Let’s pick variable xm as the dependent one. We choose such that f g 0 xm xm This allows us to rewrite the previous equation in the form f g x j j m x j n d x j 0. Because all the variables in this equation are independent, we can vary them arbitrarily, so we conclude f g 0 for all j m x j x j Combined with the equation specifying , we have f g 0 for all j x j x j Notice that Lagrange’s method doesn’t tell us how to determine . The Boltzmann Factor Maximize W (a1 , a2 , ) subject to the constraints a j A , j ln W a a E k k k 0, a j k k a E j j j j 1, 2, where and are Lagrange multipliers. Using the expression for W, applying Stirling’s approximation and evaluating the derivative we find aj e Ej E It can be shown that 1/ kBT At a temperature T the probability that a system is in a state with quantum mechanical energy Ej is Pj Q e j Ej e Ej Q ( 1/ k BT ) canonical partition function Thermodynamic Properties of the Canonical Ensemble Postulate: The ensemble average 1 P E Q j j e j Ej E j Q 1E U ( N ,V , T ) j is the observable “internal” energy. From the above, ln Q 2 ln Q U k T B T N ,V N ,V 2 1 U ln Q U 2 cv kB constant-volume heat capacity 2 2 kBT N ,V T N ,V N ,V Separable Systems The partition function for a system of two types of noninteracting particles, described by the Hamiltonian Hˆ Hˆ (1) Hˆ (2) with energy eigenvalues (2) E jk (1) j k is Q Q(1)Q(2) If the energy can be written as a sum of various (single-particle-like) contributions, the partition function is a product of the corresponding components. Distinguishable vs. Indistinguishable Particles The partition function for a system of N distinguishable particles is Q qN where q is the partition function of one particle. The partition function for a system of N indistinguishable particles is Q q N /N ! Partition Function for Polyatomic Molecules The Hamiltonian of a molecule is often approximated by a sum of translational, rotational, vibrational and electronic contributions: Hˆ Hˆ trans Hˆ rot Hˆ vib Hˆ elec Within this approximation the molecular partition function is q q trans q rot q vib qelec Translational Partition function Atom in box of volume V: 3 2 q trans 2 mkBT (V , T ) V 2 h Translational energy of an ideal gas: U trans (V , T ) 3 RT 2 Translational contribution to the heat capacity of an ideal gas: cvtrans 3 R 2 Electronic Partition function There is no general expression for electronic energies, thus one cannot write an expression for the electronic partition function. However, electronic excitation energies usually are large, so at ordinary temperatures qelec (V ,T ) 1 Vibrational Partition Function for Diatomic Molecule vvib v 12 (harmonic oscillator approximation) q vib e /2 e vib / 2T , vib "vibrational temperature" 1 e 1 e vib / T kB Vibrational energy of diatomic molecule: 1 1 U vib N 2 e 1 Vibrational contribution to heat capacity of diatomic molecule: evib / T vib R T 1 evib / T 2 cvvib As T , cvvib R 2 Rotational Partition Function for Diatomic Molecule Jrot q 2 rot J ( J 1) (rigid rotor approximation, (2J +1)-fold degeneracy) 2I 8 2 Ik BT T h2 , rot 2 "rotational temperature" 2 h rot 8 Ik B Rotational energy of diatomic molecule: U rot NkBT Rotational contribution to heat capacity of diatomic molecule: cvrot R Symmetry factors: If there are identical atoms in a molecule some rotational operations result in identical states. We introduce the “symmetry factor” to correct this overcounting. q rot T rot For homonuclear diatomic molecules at high temperature =2. Polyatomic Molecules n atoms, 3n degrees of freedom. Nonlinear molecules: 3 Translational degrees of freedom 3 Rotational degrees of freedom 3n-6 Vibrational degrees of freedom Linear molecules: 3 Translational degrees of freedom 2 Rotational degrees of freedom 3n-5 Vibrational degrees of freedom q vib q vib j , j 1 3n 5 (linear) 3n 6 (nonlinear) Rotational partition function for linear polyatomic molecules q rot T h2 , rot 2 rot 8 Ik B Symmetry factor: The number of different ways the molecule can be rotated into an indistinguishable configuration. Asymmetric molecules: 1 (e.g. COS) Symmetric molecules: 2 (e.g. CO2 , HC CH) Rotational partition function for nonlinear polyatomic molecules Rotational properties of rigid bodies: three moments of inertia IA , IB , IC . I A I B IC spherical top (e.g. CH 4 ) I A I B IC symmetric top (e.g. NH3 ) I A I B IC asymmetric top (e.g. H 2O) 8 Ik BT Spherical top: q h 2 2 3 2 rot Symmetric top: q rot Asymmetric top: q rot 8 I A k BT 8 I C k BT 2 h2 h 2 2 1 2 1 2 1 2 8 I A k BT 8 I B k BT 8 I C k BT 2 2 h2 h h 2 2 2 1 2 The symmetry factor equals the number of pure rotational elements (including the identity) in the point group of a nonlinear molecule. The Normal Mode Transformation 2 ˆ p Hˆ i +V (r1 ,r2 , i 1 2mi 3n ) (Cartesian atomic coordinates) Expand the potential in a Taylor series about the minimum through quadratic terms: V 1 3n 3n 1 T x K x x K x ( xi =ri - ri min Cartesian displacement coordinates) i ij j 2 i 1 j 1 2 2V where Kij xi x j force constant matrix We will show in a simple way how one can obtain an independent mode form by doing a coordinate transformation. In practice, the normal mode transformation proceeds after the Hamiltonian in expressed in internal coordinates. Transform to mass-weighted Cartesian coordinates qi mi xi 3n pi2 1 2 1 2 1 3n 3n Then = pi and H pi qi Kij q j 2m 2 2 i 1 j 1 i 1 2 2 2V 12 12 V Kij mi m j qi q j xi x j Introduce normal mode coordinates Qi (with conjugate momenta Pi ): U Q q q K q QT UT K U Q QT Λ Q K U UΛ UT K U Λ, or U is the orthogonal matrix of eigenvectors, L is the diagonal matrix of eigenvalues. 3n 1 2 1 3n 2 2 Now H Pi i Qi 2 i 1 i 1 2 The Equipartition Principle Every quadratic term in the Hamiltonian of a system contributes ½ kBT to the internal energy U and ½ kB to the heat capacity cv at high temperature. 2 ˆ 1 p 1 ˆ Translation in one dimension: H (one quadratic term) k B 2 m 2 2 1 d 1 ˆ Rotation about an axis: H (one quadratic term) k B 2 2 d 2 (linear molecules: 2 such terms, nonlinear molecules: 3 such terms) 2 ˆ 1 p 1 Vibration: Hˆ m 2 xˆ 2 (two quadratic terms) k B 2 m 2 Diatomic molecule: 3½ kB Linear triatomic molecule: 6½ kB Nonlinear triatomic molecule: 6 kB THE FIRST LAW OF THERMODYNAMICS The first law is about conservation of energy (in the form of work and heat) Mechanical Work Expansion of a gas removing pins m h piston held down by pins Pf , V f Pi , Vi Work performed by the gas: w Pext V Infinitesimal volume change: d w Pextd V Mechanical work: Vf w Pext (V )dV Vi Convention: work done on the system is taken as positive. Reversible Processes A process is called reversible if Psystem= Pext at all times. The work expended to compress a gas along a reversible path can be completely recovered upon reversing the path. When the process is reversible the path can be reversed, so expansion and compression correspond to the same amount of work. To be reversible, a process must be infinitely slow. A process is called reversible if Psystem= Pext at all times. Vf w P(V )dV Vi Reversible Isothermal Expansion/Compression of Ideal Gas P P Pi w nRT ln Pf V Vi Vf Reversible isothermal compression: minimum possible work Reversible isothermal expansion: maximum possible work Vf Vi Exact and Inexact Differentials A state function is a property that depends solely on the state of the system. It does not P depend on how the system was brought to that state. When a system is brought from an initial to a final state, the change in a state function is independent of the path followed. An infinitesimal change of a state function is an exact differential. Internal energy U : state function dU : exact differential i f dU U f U i U , independent of the path Work and heat are not state functions and do not correspond to exact differentials. Of the three thermodynamic variables, only two are independent. It is convenient to choose V and T as the independent variables for U. The First Law The sum of the heat q transferred to a system and the work w performed on it equal P the change U in the system’s internal energy. U q w Postulate: The internal energy is a state function of the system. Work and heat are not state functions and do not correspond to exact differentials. PdV dU dq Work and Heat along Reversible Isothermal Expansion for an Ideal Gas, where U=U(T) P P reversible constant-pressure A PA B reversible isothermal C PC V VA VC VC VA V nRTA ln C VA TB TA wAC wAB PA VB VA U AC 0 qAC nRTA ln TC qBC U BC cV dT TB TB U AB cV dT TA q AB U AB wAB cV dT PA VB VA TB TA wBC 0 VC VA Free Expansion P Suddenly remove the partition No work, no heat! U 0 U dU cV dT dV V T U U (T ) T 0 U For real, non-ideal gases these hold approximately, and is small. V T Adiabatic Processes A process is called adiabatic if no heat is transferred to or out of the system. U wad , dU dwad P P PA dU cV dT PdV reversible constant-pressure A PC VA B reversible isothermal nRT dV (ideal gas) V C If cV(T) is known, this can be used to determine T (and thus also P) as a function of V. D For a monatomic ideal gas, V VC reversible adiabatic cV 23 nR independent of T 3 2 TD VA TA VD Gases heat up when compressed adiabatically. (This is why the pump used to inflate a tire becomes hot during pumping.) Adiabatic cooling! Enthalpy H U PV P VdP enthalpy function dH dU PdV VdP dq Heat capacity at constant pressure: Ideal gas: H cP = T P cP cV nR Heat transferred at constant pressure is enthalpy change. Reversible Adiabatic Expansion of Ideal Gas Revisited c dT PdV c dT VdP 0 dq P V cV dT PdV , cP dT VdP cP d ln V d ln P cV If cP / cV is constant (e.g., monatomic ideal gas), For a monatomic ideal gas, 5 3 cP PV const. 5 5 nR, 2 3 V2 P1 P2 V1 ENTROPY AND THE SECOND LAW Processes evolve toward states of minimum energy and maximum disorder. These two tendencies are in competition. The second law is about entropy and its role in determining whether a process will proceed spontaneously. Entropy A statement of the second law: P No process is possible whose sole effect is the absorption of heat from a reservoir and the conversion of this heat into work. Postulate: There exists a state function S called the entropy. This is such that, for a reversible process, dq dS T dS 0 . 1/T is the integrating factor for dq S has units of R (or kB). For a reversible process, dU TdS PdV Isolated system is a system that cannot exchange any matter or energy with the environment. PThe second law: The entropy of an isolated system never decreases. A spontaneous process that starts from a given initial condition always leads to the same final state. This final state is the equilibrium state. Entropy of an Ideal Gas dU cV dT TdS PdV TdS nRT dV V dT dV nR T V dS cV T V T0 V0 S cV (T ) d ln T nR d ln V f (T ) f (T0 ) nR ln S (T ,V ) V V0 independent of the path! nRT dH cP dT TdS VdP TdS dP P dT dP dS cP nR T P T P T0 P0 S cP (T ) d ln T nR d ln P g (T ) g (T0 ) nR ln S (T , P) P P0 The Clausius Principle The Clausius principle states that No process is possible whose sole result is the transfer of heat from a cooler body to a hotter body. The Clausius principle is another statement of the second law. dU dU A dU B 0 (isolated system) A B dS dS A dS B 1 dU A dU B 1 dU A TA TB TA TB According to Clausius’ principle, if TA > TB then heat will flow from A to B, i.e., dS 0 A spontaneous process evolves in the direction of increasing entropy. Reversible vs. Spontaneous (Irreversible) Processes 0. In an isolated system dq dq Reversible process: dS . T dq Spontaneous (irreversible) process: dS . T dq In general, dS . T A reversible adiabatic process is an isentropic process, dS = 0. The Caratheodory Principle This is yet another statement of the second law. It states that In the neighborhood (however close) of any equilibrium state of a system (of any number of thermodynamic coordinates) there exist states that cannot be reached by reversible adiabatic processes. Caratheodory’s statement is equivalent to the existence of the entropy function. P S1 S3 S2 V Family of isentropic (constant S) surfaces that don’t intersect. Proof of Existence of Non-Intersecting Adiabatic Surfaces Suppose B can be reached from A by a reversible adiabatic process. Let’s suppose C can also be reached from A via a reversible adiabatic process. Consider the process A B C A. P A U A B C A 0 qB C wA B ,C A qB C 0 (heat absorption) wA B , C A q B C 0 C B V So in this cycle there is heat absorbed that is converted into work. This is in contradiction with the second law. We arrived at this contradiction by assuming there are two reversible adiabatic processes starting from point A. The Carnot Cycle P AB: reversible isothermal at temperature T1 A BC: reversible adiabatic B CD: reversible isothermal at temperature T2 < T1 DA: reversible adiabatic D U ABCDA 0 q AB qCD w 0 C V (the system does work) Efficiency of Carnot engine: Entropy changes: 1 S AB T2 1 (unless T2 0) T1 B A w q 1 CD q AB q AB dq q q AB , SCD CD S AB T T1 T2 qCD T 2 q AB T1 One can never utilize all the thermal energy given to the engine by converting it into mechanical work. The Internal Combustion Engine In the gasoline engine, the cycle involves six processes, four of which require motion of the piston and are called strokes. The idealized description of the engine is the Otto cycle. 1. Intake stroke. A mixture of gasoline vapor and air is drawn into the cylinder (EA). 2. Compression stroke. The mixture of gasoline vapor and air is compressed until its pressure and temperature rise considerably (AB). 3. P C Ignition. Combustion of the hot mixture is caused by an electric spark. The resulting combustion products attain a very high pressure and temperature, but the volume remains unchanged (BC). 4. Power stroke. The hot combustion products expand and push the piston out, thus expanding adiabatically (CD). 5. Valve exhaust. An exhaust valve allows some gas to escape until the pressure drops to that of the atmosphere (DA). 6. Exhaust stroke. The piston pushes almost all the remaining combustion products out of the cylinder (AE). D B E A V Thermodynamics of the Otto Cycle P Reversible adiabatic compression AB: TAVA 1 TBVB 1 C BC is reversible absorption of heat qh from a series of D reservoirs whose temperatures range from TB to TC : TC qh cV dT . B TB E If we assume cV is constant, qh cV (TC TB ). A V Reversible adiabatic expansion CD: TCVC 1 TDVD 1 or TCVB 1 TDVA 1 DA is reversible rejection of heat qc to a series of reservoirs whose temperatures range TA from TD to TA : qc cV dT cV (TD TA ). TD 1 q V T T 1 c 1 D A 1 B qh TC TB VA VB / VA : compression ratio Other Ideal Gas Engines See http://www.ac.wwu.edu/~vawter/PhysicsNet/Topics/ThermLaw2/Entropy/GasCycleEngines.html copied in 444-web-page/Ideal Heat Engine Gas Cycles.htm Entropy of Reversible Isothermal Expansion of an Ideal Gas Reversible isothermal expansion: dq sys dU sys d wsys But dU sys 0 dq sys PdV Ssys 2 1 dq sys T V2 V1 V2 dV PdV V2 nR nR ln 0 V1 V T V1 The heat entering the system was absorbed from the environment. Then dq Senv Ssys , S univ 0. env dq sys Entropy of Spontaneous Expansion of an Ideal Gas Spontaneous expansion: q sys U sys wsys 0 Entropy is a state function, so the entropy change of the system has the same value as that during a reversible (isothermal) expansion: Ssys V2 nR ln 0 V1 Because no heat is absorbed from the environment, dq Senv 0, S univ 0. env 0 The entropy of the system increased, but the entropy of the environment remained unchanged. Statistical Mechanical Definition of Entropy Sensemble kB ln W Completely ordered ensemble: Maximum disorder: a1 1, a2 a3 a1 a2 a3 Ssys 0 Sensemble 0 (set that maximizes W ) 1 Sensemble A Ssys 1 Sensemble A A ! Use W and apply Stirling's approximation. a ! j j ln W lnA ! ln a j ! A lnA A a j ln a j a j j j Sensemble k BA lnA k B a j ln a j j Use populations p j Sensemble aj A k BA lnA k B A p j lnA p j j k BA lnA k BA p j lnA k BA j p j Ssys k B p j ln p j j j ln p j Pure and Mixed States If all replicas of our system in a particular ensemble are in the same state n, i.e., pn 1, pi n 0 then S 0. This is called a pure ensemble. Note: the quantum state n need not be an eigenstate of the Hamiltonian. If the members of the ensemble are in different quantum states, i.e., pi 1 for all i, then S 0. This is called a mixed ensemble. The canonical ensemble is a mixed ensemble. Entropy of the Canonical Ensemble pi Q 1e Ei S k B Q 1 e Ei Ei ln Q k B U k B ln Q i S U k B ln Q T ln Q From U k BT it follows that T N ,V 2 ln Q S k BT k B ln Q T N ,V Entropy of Monatomic Ideal Gas ln Qtrans S k BT k B ln Qtrans T N ,V 3N ln Qtrans . Using Stirling's approximation to ln N !, T N ,V 2 T 2 mk T 2 V 5 B S nR nR ln 2 2 N A h 3 Molecular Interpretation of Work and Heat U pjEj, j pj e Ej Q dU p j dE j E j dp j j j variation of energy levels without changing populations; can be done by changing the volume. change populations without changing energy eigenvalues; can be done by heating or cooling E j dU p j dV E j dp j PdV dq j j V N Q E E j E j Q e j , e V N , j j V N E j ln Q P p j k BT V V N , j N E dp dq j j j Example: Monatomic ideal gas 3N 2 Q 1 2 m N V N ! h2 N T P k BT nR V V N ln Q V N , V ideal gas law! We see that the ideal gas law is obtained by using relations obtained for a gas of non-interacting particles. The Boltzmann Factor: Determination of the Lagrange Multiplier S kB p j ln p j dS kB ln p j dp j dp j kB ln p j dp j j because j dp j j 0 j dq dS kB E j ln Q dp j kB E j dp j kB dq T j j kB 1 T THE THIRD LAW The third law is about the impossibility of attaining the absolute zero of temperature in a thermodynamic system. Entropy as a Function of Temperature U S cV T T V T V P dU TdS PdV T2 S (T2 ) S (T1 ) cV (T )d ln T under constant V T1 H S dH TdS VdP c T P T T P P T2 S (T2 ) S (T1 ) cP (T )d ln T under constant P T1 The third law: Absolute zero is not attainable via a finite series of processes. P according to the Nernst-Simon statement, or, The entropy change associated with any isothermal reversible process of a condensed system approaches zero as the temperature approaches zero. dS cV dT at constant V T Crystals: cV T 3 as T 0 dS T 2dT as T 0 S kB p j ln p j j The entropy of a system that has a non-degenerate ground state vanishes at the absolute zero. First-Order Phase Transitions P Many thermodynamic variables are discontinuous across first-order phase transitions. S Phase I T T0 T 0 H T0 cPI (T )dT T0 cPII (T )dT (constant P) Phase II T Helmholtz and Gibbs “Free” Energies A U TS Helmholtz free energy dA TdS PdV TdS SdT SdT PdV A A(T ,V ) G A PV U TS PV Gibbs free energy dG SdT PdV PdV VdP SdT VdP G G (T , P ) Reversible isothermal process under constant volume: dA = 0 Reversible isothermal process under constant pressure: dG = 0 Legendre Transforms It is often desirable to express a thermodynamic function in terms of different independent variables. Most often this new desirable variable is the first derivative of a fundamental function with respect to one of its undesirable independent variables; for example, U U ( S ,V ) U dU TdS PdV , P V S We are seeking a general tool for finding a new function that contains the same information as the original fundamental thermodynamic function, but where the “undesirable” variable has been eliminated in favor of the “desirable” one. In the previous example, We seek a new function H H ( S , P) that is equivalent to U but which depends on the variable P rather than V . General Theory of Legendre Transformation Given a function f (x), we seek an equivalent function (i.e., one containing the same information as f ) whose independent variable is df /dx. f The curve f (x) can be reconstructed from the family of its tangent lines. x A tangent line can be specified by its slope f (new independent variable) and intercept . f Notice f f x f ( x) “Legendre tranform of f” x We solve f ( x) for x and substitute in the above relation to obtain ( x). This is possible if f is single-valued, i.e., f 0 at all x. Application of Legendre Transform Theory U U ( S ,V ) dU TdS PdV U T S V U P V S H H ( S , P) U ( P)V dH TdS VdP A A(T ,V ) U TS dA SdT PdV A P V T G G (T , P) A ( P)V H T S P H TS U TS PV dG SdT VdP Maxwell’s Relations f ( x1 , x2 ) df y1dx1 y2 dx2 f y1 x1 x2 f y2 x2 x1 y2 y1 x x 1 x2 2 x1 Conversely, if the Maxwell relation is satisfied, one can conclude that df is an exact differential. Examples dU TdS PdV U T S V U P V S T P V S S V dA SdT PdV A A S , P T V V T S P V T T V dH TdS VdP dG SdT VdP T V P S S P S V P T T P Applications of Maxwell’s Relations Entropy of a gas: S S dS dT dV T V V T S P At constant T , dS dV dV V T T V V2 P S 2 S1 dV along an isothermal process V1 T V nR P Ideal gas: T V V S 2 S1 nR ln V2 V1 Internal energy of a gas: dU TdS PdV U S P P T P T V T V T T V U P At constant T , dU dV P T dV V T T V V2 P U 2 U1 P T dV along an isothermal process V1 T V We may choose a sufficiently large value of V1 such that U1 is given by the ideal gas law, then calculate the internal energy at a different volume where the gas does not exhibit ideal behavior. Pfaffian Forms Extensive variable Intensive variable Work V (gas volume) -P -P dV L (wire length) F (force) F dL S (surface tension) -S dA A (film area) M (magnetic dipole moment) H (magnetic field) … … dU TdS Yi dX i Pfaffian form i X i : extensive variable Yi : intensive variable H dM … PHASE EQUILIBRIUM What is the equilibrium state of a multi-component system? Phase Diagrams Water Carbon Dioxide (Typical Case) Phase transitions (melting, freezing, boiling, sublimation, etc.) Chemical Potential Intensive variable m such that dw "chemical work" chem m j dn j j (The sum is over all components of a system and nj are the mole numbers.) dU TdS PdV m j dn j j dH TdS VdP m j dn j j dA SdT PdV m j dn j j U H mi n n i i S ,V ,n S , P ,n j i j i A G n n i T ,V ,n j i i T , P ,n j i dG SdT VdP m j dn j j Pure substance: G( P, T , n) n g ( P, T ) m g ( P, T ) The chemical potential of a pure substance is the molar Gibbs free energy. General Conditions of Equilibrium An isolated system tends to attain the state of maximum entropy with respect to its internal (extensive) degrees of freedom, subject to the given external constraints. dS = 0, d2S < 0 Consequence: The thermodynamic potentials attain minimum values with respect to their internal extensive variables at equilibrium, subject to the given external constraints. I. Thermal Equilibrium Constraints: A B V A const., V B const., U U A U B const. "Internal variable": U A rigid, diathermal wall impermeable to matter U A U A ( S A ,V A ) and S S A (U A ,V A ) S B (U B ,V B ) Since the volumes cannot change, S A S A S A S A A B A dS dU dU dU A B A B U V A U V B U V A U V B 1 S From dU TdS PdV we find and therefore U T V 1 1 dS A B dU A T T A B At equilibrium dS 0 for any dU A . It follows that T T II. Thermal and Mechanical Equilibrium Constraints: A B V V A V B const., U U A U B const. "Internal variables": U A , V A movable, diathermal wall impermeable to matter S A S A S A S A B B A A dV dU dV dU dS B B A A V U B U V B V U A U V A S A S A S A A S A A B dV dU A B A V U A V U B U V A U V B P 1 S S and therefore , From dU TdS PdV we find T V T U U V P A PB A 1 1 A dS A B dU A B dV . T T T T At equilibrium dS 0 for any dU A , dV A . It follows that T A T B , P A PB III. Equilibrium with Respect to Matter Flow A Rigid, diathermal wall permeable to substance 1 B Internal variables: U A , n1A S A S B S A S B A A B B dS dU dn dU dn 1 1 A B A B U n U n A A B B A A B B V ,n1 V ,n1 1 V ,U 1 V ,U dU TdS PdV m j dn j j S m1 n T 1 U ,V m1A m1B A 1 1 A dS A B dU A B dn1 T T T T At equilibrium T A T B , m1A m1B 1st Order Phase Transitions: The Clausius-Clapeyron Equation P Phase I B A A Phase II T d m I m BI m AI S I dT V I dP d m II m BII m AII S II dT V II dP V II V dP S S dT I II I dP H dT T V dP S III dT V III Clapeyron equation Liquid-to-vapor transition S l g 0, V l g 0 dP 0 dT Increase in pressure causes conversion to the higher-density liquid phase. Solid-to-liquid transition S s l 0 If V s l 0 then If V s l 0 then dP 0 dT dP 0. This is the case with water. dT The Clapeyron equation is an expression of Le Chatelier’s principle. Approximation for liquid-vapor phase transition: Vg V l, V g RT (ideal gas), so P 1 dP H l g P dT RT 2 Clausius-Clapeyron approximation Statistical Mechanical Calculation of Chemical Potential G A n P ,T n T ,V m ln Q ln Q A U TS kBT T k BT k B ln Q T T N ,V N ,V 2 A k BT ln Q ln Q ln Q RT n T ,V N T ,V m k BT Chemical Potential of Ideal Gas qN Q ln Q N ln q ln N ! N ln q ( N ln N N ) N! ln Q q ln N N n PV nRT N A RT Nk BT NA m RT ln V k BT N P q k BT q RT ln k BT RT ln P V P V q kBT P m RT ln RT ln , or 0 V P P0 P0 standard pressure (105 Pa) P m m0 RT ln P0 SOLUTIONS We consider a two-component system with mole numbers n1 and n2. dG VdP SdT m1dn1 m2 dn2 dG m1dn1 m2 dn2 at constant P and T Imagine increasing the mole numbers from 0 to their final values by varying a dimensionless parameter : dn1 n1d , dn2 n2 d n1 n2 1 0 0 0 G m1dn1 m 2 dn2 ( m1n1 m 2 n2 )d m1n1 m 2 n2 G G1 G2 G1n1 G2 n2 Gi : partial molar free energies Gi mi The partial molar free energy is the chemical potential of the substance, i.e., an intensive variable. However, the partial molar free energies generally depend on the mole fraction n1 /(n1 n2 ) . This is so because the partial derivatives G mi ni T , P ,n j i are functions of all the variables, including nj. Using a similar procedure we can write V V1 V2 V1n1 V2n2 (Vi : partial molar volumes) The partial molar volumes depend on the mole fraction of the particular substance in the solution and are not additive when substances are mixed! This statement applies generally to any extensive variable. Of course extensive variables still scale linearly with the total number of moles, provided the mole fraction of each substance remains fixed. Euler Relations The internal energy U is a function of extensive variables, U U (S ,V , n1 , n2 , ). Based on the previous remarks, U is a homogeneous first order property, i.e., U ( S , V , n1 , n2 , ) U ( S ,V , n1 , n2 , ) Differentiating with respect to , U ( S , V , n1 , n2 , ) U ( S , V , n1 , n2 , ) ( S ) U ( S , V , n1 , n2 , ) (V ) ( S ) (V ) U ( S , V , n1 , n2 , ) ( n1 ) ( n1 ) U ( S ,V , n1 , n2 , ) This is true for any value of . For = 1, U ( S , V , n1 , n2 , ) U ( S ,V , n1 , n2 , ) U U U S V n1 S V n1 U U TS PV mi ni i This is called Euler’s relation for the internal energy. Other Euler relations: H U PV H TS mi ni i A U TS A PV mi ni i G A PV G mi ni i The Gibbs-Duhem Relation Differentiating the Euler relation for dG, dG m j dn j n j d m j j j Using the relation dG SdT VdP m j dn j j we obtain the Gibbs-Duhem relation SdT VdP n j d m j 0 j This result can also be obtained from the Euler relation for dU: dU TdS SdT PdV VdP m j dn j n j d m j j j Using the relation dU TdS PdV m j dn j j we find SdT VdP n j d m j 0 j For a one-component system we recover the known result SdT VdP nd m dG Phase Equlibrium in Multicomponent Systems n1g , n2g n1l , n2l Two-component liquid at equilibrium with its vapor. At constant P and T, G G l dG l dn1 g dn1g n1 P ,T ,n2l ,n1g ,n2g n1 P ,T ,n1l ,n2l ,n2g G G l l dn2 g dn2g n2 P ,T ,n1l ,n1g ,n2g n2 P ,T ,n1l ,n2l ,n1g Since dn1l dn1g 0, dn2l dn2g 0, and G l m , etc. 1 l n1 P ,T ,n2l ,n1g ,n2g m1l m1g , m2l m2g Assuming the vapor behaves as an ideal gas, the chemical potential of substance j in the solution is m m m (T ) RT ln l j g j 0 j Pj P0 For the pure substance j, m lj* m gj * m 0j (T ) RT ln Pj* P0 because m 0j (T ) doesn't depend on mole fractions. It follows that m m RT ln l j l* j Pj Pj* Ideal Solutions and Raoult’s Law If the partial vapor pressure of each component in a solution obeys the relation Pj x j Pj* where x j nj ni is the mole fraction of component j in the liquid phase, i the solution is called ideal. Ideal solutions follow Raoult’s law, * m sol m j j RT ln x j Here m sol j is the chemical potential of (liquid) component j in the solution and m *j is the chemical potential of the pure substance. Vapor Pressure of Ideal Two-Component Solutions P P1 P2 x1P1* x2 P2* x1P1* (1 x1 )P2* x1 (P1* P2* ) P2* P * 1 P P P1 P2 x1 The mole fraction of component 1 in the liquid phase is P P2* x1 * P1 P2* P2* 0 (linear in x1 ) 1 Calculate the mole fraction y1 of component 1 in the vapor phase at a given value P of the vapor pressure using Dalton’s law of partial pressures: P1 y1 P (Dalton's law) x1 P1* P P2* P1* P1* P2* * * 1 * * * * x1 P1 x2 P2 P1 P2 P P1 P2 P liquid-vapor coexistence x1 depends linearly on P. P vs. x1 y1 depends nonlinearly (hyperbolically) on P (and on x1 )! P1* P vs. y1 P2* 0 x1 or y1 1 How much liquid vs. vapor is there at a pressure PC, given that the overall mole fraction of component 1 is x1B ? Mole fractions in liquid and vapor phases: n1l n1l x l l, l n1 n2 n n1g n1g y g g g n1 n2 n E 1 n1l n1g x l n ng B 1 n x x l B 1 E 1 F 1 or n1l nl x1E , n1g n g y1F x1B nl n g n1l n1g nl x1E n g y1F n y g F 1 x B 1 A "lever rule" P vs. x1 * 2 P P1* B E C F D P vs. y1 0 x1 or y1 1 Non-Ideal Solutions Pj x j Pj* P P1* P P P1 P2* P1* P1 P2* P2 P2 0 1 x1 attractive interactions between different molecules dominate Pj 0 x1 repulsive interactions between different molecules dominate x j Pj* as x j 1 only 1 Temperature-Composition Diagrams P vapor T1* Tb vs. y1 T2* Tb vs. x1 liquid 0 1 Substance labeled 2 is assumed to have a lower boiling point. The vapor is richer than the solution in the more volatile substance 2, thus y1 < x1.. Fractional distillation exploits this principle. Azeotropes P vapor T1* Tb vs. y1 T2* Tb vs. x1 liquid 0 x1 1 Fractional distillation cannot separate the two components. Activity For any solution (ideal or not), m m RT ln l j For ideal solutions Pj * j P For nonideal solutions l* j Pj Pj* xj Pj * j P a j "activity of component j in the solution" m lj m lj* RT ln a j Solid-Liquid Solutions A B A: water B: water + sugar Solutions separated by membrane permeable to water only. m Aj m Bj for every substance that appears on both sides and m wA m w* m wB m w* RT ln aw These cannot be equal with aw 1 unless m wA m wB . The only way for this to happen is to have P A P B . Osmotic Pressure m wA (T , P) m w* (T , P) B A m wB (T , P ) m w* (T , P ) RT ln aw m w* Pw * aw * . But V w , so Pw P T m w* (T , P ) m w* (T , P) Assuming Vw is independent of pressure for the liquid, P P Vw ( P)dP Vw so Vw RT ln aw 0 P P Vw ( P)dP For a dilute solution aw xw 1 xs ln aw ln(1 xs ) xs Vw RTxs xs ns nw Vw RTns It follows that the osmotic pressure of dilute solutions is given by the relation cRT Additional Definitions Coefficient of Thermal Expansion 1 V V T P Isothermal Compressibility Factor 1 V T V P T KINETIC THEORY OF GASES Root-Mean-Square Velocity u ux , u y , uz velocity vector of a gas molecule p2 3 kBT 2m 2 From the equipartition principle, u 2 1 2 urms 3kBT m Velocity Distribution Recall that the probability of having translational energy E 1 m u x2 u y2 u z2 2 is given by the Boltzmann factor P( E ) e m ux2 u 2y uz2 / 2 k BT The probability of having a velocity component ux in the x direction is Gaussian: m mu x2 / 2 k BT px (u x ) e 2 k BT From this, u x 0 and u 2 x u x2 px (u x ) k BT m The Maxwell-Boltzmann Distribution Calculate the probability distribution f (u) for a molecule to have a velocity modulus u u u u 2 x 2 y 2 z 1 2 by converting to spherical polar coordinates and integrating over angles: 2 0 0 f (u )du u du sin d 2 3 2 m mu 2 / 2 kBT d e 2 k BT 3 2 m 2 mu 2 / 2 kBT f (u ) 4 ue 2 k BT Average speed: u u f (u )du 8kBT m u 8kBT m Most Probable Speed The maximum of the Maxwell-Boltzmann distribution lies at d f (u ) 0 du which is satisfied for ump 2 k BT m Velocity Distribution and Reaction Rates The shape of the Maxwell-Boltzmann distribution has important implications for chemical reactions. Even though the maximum of the curve depends weakly on temperature, the fraction of molecules with velocities higher than a critical value depends exponentially on the temperature. Thus a relatively small increase of temperature can have a large effect on the rate of a chemical reaction. REACTION RATES Chemical Reactions A A BB CC D D 1 d [A] 1 d [B] 1 d[C] 1 d[D] A dt B dt C dt D dt k[A] [B] Exponents: reaction order d [A] k[A] [A] [A]0 e kt dt d [A] 1 1 2 Second order reactions: k[A] kt dt [A] [A]0 First order reactions: 1st Order Reactions A k k d [A] d [B] k [A]+k [B] dt dt Steady state (dynamic equilibrium): [B]eq d [A] k 0 K dt [A]eq k At all times, [A] [B] [A]0 B k [A]eq k [B]eq k [A]0 [A]eq k k [A]eq k [A]0 At all times, [A] [B] [A]0 Steady state (dynamic equilibrium): [B]eq d [A] k 0 K dt [A]eq k k [A]eq k [B]eq k [A]0 [A]eq k k [A]eq k[A]0 d [A] k [A] k [A]0 [A] k k [A] k k [A]eq dt d [A] k k [A] [A]eq dt d [A] [A]eq k k [A] [A]eq dt [A] [A]eq [A]0 [A]eq e ( k k )t Temperature Dependence of Rate Constants In most cases under common conditions, k A e Ea / RT Arrhenius equation Ea : activation energy reactants products reaction coordinate Eb Transition State Theory Assumption: All trajectories that reach the barrier top lead to products. k TST Numerator = p d ( x xb ) ( p) m dp e H ( x , p ) dx dp e H ( x , p ) 0 dp e dx H ( xb , p ) p e Eb p 2 /2 m Eb p dp e e 0 m m Denominator = Qrcl (partition function of reactants) Assuming the potential is harmonic about the minimum, cl r Q dx dp e p2 1 m02 ( x x0 )2 2 m 2 k TST 2 0 0 E e 2 b Taking into account degrees of freedom orthogonal to the reaction coordinate, † k TST k BT Q † Eb e h Qr † Q † : partition function of stable modes at the transition state Qr : partition function of reactants Corrections to Classical Transition State Theory 1. 2. Recrossing of transition state region (negative corrections) Quantum mechanical effects (primarily tunneling) ln k activated crossing tunneling regime 1/T Tunneling Preliminaries P(E) classical 1 E ? quantum mechanical Vb 0 Vb E Ptunneling (E) exp 1 2m[V (x) E]dx • Tunneling is important in the kinetics of light particles (primarily e-, H, H+, D,…) • Tunneling effects are sensitive to isotopic substitution Time evolution in double wells • Symmetric double well in a dissipative medium: quenched tunneling oscillations 1 very small splittings • Isolated symmetric double well: constant amplitude tunneling oscillations • Asymmetry quenches tunneling 0.5 0 -0.5 -1 0 4 8 t 12 16 Finite temperature reaction rates En / kBT Generally, Pc (T ) e n Pmc(En) microcanonical thermally averaged Arrhenius rate plot Classical rate theory: V / kBT b Tunneling dominates at low temperatures, where the classical rate goes to zero. log k kcl(T ) Ae kqm kcl 1/T Early history 1927 Hund suggested that quantum mechanical tunneling may play an important role in some chemical reactions. 1928 Fowler and Nordheim observed velocities of electrons emitted by metals that were too small. e- V(r) r R. H. Fowler and L. Nordheim, Proc. Roy. Soc. A 119, 173 (1928) 1932 The discovery of deuterium provided ample evidence for quantum tunneling and motivated theoretical and experimental work on isotope effects. p+ n p+ 1933 Kinetic information from ortho/para-H2 interconversion revealed considerable deviations from Arrhenius behavior indicative of tunneling 1934 Observation of tunneling splittings in NH3 1956 H+/D+ abstraction from 2-ethoxycarbonylcyclopentanone Chemical bonding, conjugated systems and band structure V(r) R * b R conduction valence Electron tunneling in biomolecules Quantum paths for the tunneling electron in ruthenium-modified myoglobin. The heme and ruthenium redox centers are separated by 28 Angstroms. KA. Kuki A. and P. G. Wolynes, Science 1987, 236, 1647-1652. Exciton tunneling in molecular aggregates Exciton tunneling in symmetric molecular aggregates leads to a type of band structure and delocalized states. Nuclear tunneling in electron transfer Fe2+ Fe3+ + e- R. A. Marcus, 1992 Nobel Prize in Chemistry. Tunneling of atoms scattering bimolecular reactions predissociation unimolecular decay enhancement of tunneling asymmetric isomerizations symmetric isomerizations Tunneling effects in bimolecular reactions A+BC AB+C H+H2 D+H2 H2 +H HD+H RBC (observed through interconversion of ortho- and para-forms) Potential surface curvature (“corner cutting”) RAB Tunneling effects in molecular spectroscopy Tunneling leads to splitting of rovibrational levels in symmetric isomerizations. The splitting is observed spectroscopically in the microwave region. Inversion of NH3 1.3cm1 C. E. Cleeton and N. H. Williams, Phys. Rev. 45, 234 (1934). H tunneling in hydrogen-bonded molecules 3,7-dichlorotropolone Tunneling in enzymes Tunneling plays a significant role in hydrogen transfer at enzyme active sites. Motion of the primary (1°) and secondary (2°) hydrogens in the reaction of alcohol dehydrogenase. A. Kohen, R. Cannio, S. Bartolucci and J.P. Klinman (1999), Nature 399, 496-499. Tunneling in the condensed phase TST kqm kqm 5 4 4 3 V / kBT b kcl(T ) Ae 2 activated dynamics log Classical rate theory: 3 2 deep tunneling 1 0 1 0 1 / m 2 Intermediate T 3 0 1 / m Low T 2 3 Rotational tunneling in crystals At low temperatures (4-50K) the rotation of ammonium ions in ionic salts is dominated by tunneling. H. L. Strauss, Acc. Chem. Res. 30, 37-42 (1997). Competing effects in kinetics Diffusion of H and D in crystalline Si conventi onal TST TST for 1dim. adiabatic potential quant um Inverse isotope effect! Theoretical treatments of tunneling • Full solution of the quantum mechanical wave equation • Instanton theory (tunneling in imaginary time / inverted potential) • Tunneling corrections to classical trajectory calculations from semiclassical expressions • Quantum mechanical solution of simplified models (master equations, harmonic bath approximations,…) • Path integral or quantum Monte Carlo calculations in select situations (e.g., tunneling splittings) Scanning Tunneling Microscopy A tip is scanned over a surface at a distance of a few atomic diameters in a point-by-point and line-by-line fashion. At each point the tunneling current between the tip and the surface is measured. The tunneling current decreases exponentially with increasing distance and thus, through the use of a feedback loop, the vertical position of the tip can be adjusted to a constant distance from the surface. Gerd Binnig and Heinrich Rohrer, IBM Research Laboratory, Zurich, shared the Physics Nobel prize in 1986 for their discovery of STM. Imaging surfaces Unreconstructed (110) Ni surface Cu surface (electron standing waves on surface steps) Cr impurities on a Fe (001) surface Zig-zag chain of Cs atoms on the GaAs(110) surface. Nanoengineering with STM Spelling “atom” in Japanese. Fe on Cu Stadium quantum corral: Fe on Cu STABILITY CRITERIA AND PHASE TRANSITIONS Concavity of the Entropy Imagine a system whose entropy function of a system has the shape shown in the figure. Consider two identical such systems, each with internal energy U0 and entropy 2S(U0). Suppose we remove energy U from the first system and put it in the second system. The new entropy will be S (U U ) S (U U ) 2S (U ) S U0 - U U0 U0 + U U Since this rearrangement of the energy results in a larger entropy, it should occur spontaneously if the two systems are connected through a diathermal wall. This way the system will break up into two systems of different thermodynamic properties. This process is a phase transition. The instability leading to phase separation is a consequence of the assumed convex shape of S over a range of U. In stable thermodynamic systems the entropy function is a concave function, i.e., d 2 S < 0 with respect to the extensive variables U and V. Stability Conditions for Thermodynamic Potentials The concavity condition for the entropy implies the convexity of the internal energy function with respect to its extensive variables S and V, as illustrated in the figure. Adapted from H. B. Callen, Thermodynamics and an Introduction to Thermostatistics, 2nd Edition. V V The other thermodynamic potentials are functions of extensive as well as intensive variables. Because intensive variables are introduced through negative terms in the Legendre transform of the internal energy, the resulting thermodynamic potentials are concave functions of their intensive variables (but they are still convex functions of their extensive variables). For example, 2 A 2G 2 0, 2 0 V T T P A consequence of the first of these relations is 2 P A 2 0 T 0 V T V T V First Order Phase Transitions G or U Failure of stability criteria: If the fundamental thermodynamic function of a system is unstable, fluctuations may take the system over the local maximum, and the system breaks up into more than one phases. S or V Second Order Phase Transitions and Critical Phenomena The two stable minima responsible for a first order phase transition coalesce at the critical point, giving rise to a second order phase transition. G or U Critical phenomena are accompanied by huge density fluctuations, which give rise to the observed “critical opalescence”. S or V SUPERFLUIDITY AND BOSE-EINSTEIN CONDENSATION The History of Superfluid 4He 1908: 4He was first liquified (5.2 K). Unusual properties were observed: strange flow, expansion upon cooling below 2.2 K. 1928: Sharp maximum in the density with a discontinuity in slope at ~2.2 K. Two phases. 1932: The specific heat diverges at 2.17 K; the curve has a shape (“lambda transition”). Normal and superfluid phases identified. 1930s-1940s: Remarkable transport properties of superfluid 4He studied extensively. • Viscosity drops by many orders of magnitude; the system flows through capillaries. • The superfluid forms extended thin films over large surfaces. • The superfluid does not rotate upon rotating its container. • It appears the superfluid flows without friction! The Phenomenon of Superfluidity A group of phenomena including: • Frictionless flow • Persistent current • Heat transfer without a thermal gradient Bose-Einstein Condensation (BEC) Einstein predicted that if a gas of bosons were cooled to a sufficiently low temperature, all the atoms would gather in the lowest energy state. In 1995, Cornell and Wieman produced the first condensate of 2000 Rb atoms at 20 nK. Ketterle produced a condensate of Na with more atoms and observed interference patterns. BEC is intimately connected with superfluidity, but is not a necessary condition for this group of phenomena. Recent Nobel prizes: 2001: Cornell, Wieman and Ketterle for BEC 2003: Leggett for theory of superfluids Condensate fraction: n0 N0 N (N0: number of particles in the zero momentum state) In the strongly interacting 4He superfluid the condensate fraction is small (about 7% at T = 0) The Quantum-Classical Isomorphism A single quantum mechanical particle is isomorphic to a “necklace” of N classical “beads” that are connected with one another via harmonic springs and which experience a potential equal to 1/N of the actual potential felt by the quantum particle. Quantum statistical effects of identical bosons or fermions manifest themselves in the exchange of beads, which causes the necklaces of different particles to cross-link. A snapshot of 4He at 1.2 K. Each 4He atoms is represented in the simulation through 20 “pair-propagator” beads. The blue beads correspond to linked necklaces. The End