Lecture 18
advertisement

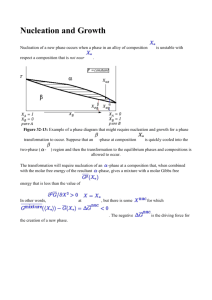
Surfaces Lecture 18 Diffusion Rates • Probability of atom making a jump to vacancy is P =Àe- EB /RT • where is number of attempts and EB is the activation or barrier energy. • Combining, total diffusion rate will be product of probability. of a vacancy times probability of a jump:  = me-EB /RT + ne-(EH +EB )/RT o where m and n are constants • At higher T, thermal vacancies dominate and  @ ne-(EH +EB )/RT • Bottom line: like other things in kinetics, we expect diffusion rates to increase exponentially with temperature. Diffusion Rates • In general, the temperature of the diffusion coefficient is written as: D = Doe- EB /RT • In log form: ln D = ln Do - EA RT • At lower temperature, where permanent vacancies dominate, we might see different behavior because:  = me-EB /RT + ne-(EH +EB )/RT Determining Diffusion Coefficients • In practice, one would do a experiments at a series of temperatures with a tracer, determine the profile, solve Fick’s second law for each T. • Plotting ln D vs 1/T yields Do and EB. • Diffusion coefficient is different for each species in each material. • Larger, more highly charged ions diffuse more slowly. Reactions at Surfaces Homogeneous reactions are those occurring within a single phase (e.g., an aqueous solution). Heterogeneous reactions are those occurring between phases, e.g., two solids or a solid and a liquid. Heterogeneous reactions necessarily occur across an interface. Interfaces, Surfaces, and Partial molar Area • By definition, an interface is boundary between two condensed phases (solids and liquids). • A surface is the boundary between a condensed phase and a gas (or vacuum). o In practice, surface is often used in place of interface. • We previously defined partial molar parameters as the change in the parameter for an infinitesimal addition of a component, e.g., vi = (∂V/∂n)T,P,nj. We define the partial molar area of phase ϕ as: æ ¶A ö aif = ç è ¶ni ÷ø T ,P,n j¹i • where n is moles of substance. • Unlike other molar quantities, partial molar area is not an intrinsic property of the phase, but depends on shape, size, roughness, etc. For a perfect sphere: a= ¶A ¶V 2v = ¶V ¶n r Surface Free Energy • We now define the Surface Free Energy as: f æ ¶G ö sf = ç ÷ è ¶A ø T ,P,n • The surface free energy represents those energetic effects that arise because of the difference in atomic environment on the surface of a phase. • Surface free energy is closely related to surface tension. • The total surface free energy of a phase is minimized by minimizing the phase’s surface area. o Thus a water-drop in the absence of other forces will tend to form a sphere, the shape that minimizes surface area. Incorporating Surface Free Energy • When surface effects must be considered, we can revise equation 3.14 to read: dG = VdP - SdT + å µ dn + ås dA • where the second sum is taken over all interfaces and surfaces of the phase. i i i k k k o In general, the surface free energy depends on the nature of the two phases in contact. It will be different for water in contact with a mineral than for water in contact with air. o For an isotropic phase immersed in a homogeneous medium, we need concern ourselves with only one interface free energy. o For crystals, different faces can have different interfacial free energies, even if in contact with the same phase (but we won’t concern ourselves with this complexity). o In a rock, for example, a given crystal might be in contact with several different minerals - each would have a different interfacial free energy. The Kelvin Effect • When the size of phases involved is sufficiently small, surface free energy can have the effect of displacing equilibrium. For an equilibrium system at constant temperature and pressure, eqn. 5.109 becomes: 0 = ån i µio + RT ån i lnai + ås k dAk • • i k The first term on the right is ∆G˚, which is equal to –RT ln K. This is the “normal” equilibrium constant, so we’ll call it K˚. We’ll call the summation in the second term Ks, the equilibrium influenced by surface free energy. Making these substitutions and rearranging, we have: s k dAk å ln K s = ln K˚ - k RT Thus we predict that equilibrium can be shifted due to surface free energy, and the shift will depend on the surface or interfacial area. This is known as the Kelvin effect. o o There are a number of examples of this effect. For example, fine, and therefore high surface area, particles are more soluble than coarser particles of the same composition. Water has a surface free energy of about 70 mJ/m2. So, for example, humidity in clouds and fogs can reach 110% when droplet size is small. Surface Free Energy & Metamorphism • One usual effect of metamorphism is an increase in grain size. This occurs even in monomineralic rocks like limestone and sandstone. • The free energy of the system is reduced by reducing grain-to-grain interfaces, which is lower in coarser grained rocks. Nucleation • Liquids can become significantly supersaturated but crystallization will often begin as soon as seed crystals are added. o o • • This suggests that nucleation is an important barrier to crystallization. This barrier arises because the formation of a crystal requires a local increase in free energy due to the surface free energy at the solid–liquid interface. Let’s explore this a bit further. For a crystal growing in a liquid, the complete free energy change is: dG = dσ+dGxtl For a spherical crystal of phase ϕ growing from a liquid solution of component ϕ. The free energy change over some finite growth interval is: 4 3 ∆ Gxt 2 ∆ Gtot = 4p r s + p r 3 • • V r is radius. We divide by the molar volume to convert J/mol to J/m3. (In fact, per volume units, rather than per mass or per mole, turn out to generally be more convenient in kinetics). The first term on the right is always positive, so exactly at saturation, ∆Gtot is positive and there will be no crystallization. Nucleation & Growth 4 ∆ Gxt ∆ Gtot = 4p r 2s + p r 3 3 V • For small r, first term increases more rapidly. • ‘Turnaround’ occurs at ∂∆G/∂r = 0 ¶∆ Gtot ∆ Gxt = 8p rs + 4p r 2 ¶r V • Setting ∂∆G/∂r = 0, we find the critical radius: rcrit -2s = ∆ Gxtl / V Total free energy as a function of r for various amounts of undercooling. We approximate the ∆G term as ∆G ≈ -∆T∆Sxtl, where ∆T is the difference between actual temperature and the saturation temperature. Surface Free Energy & Viscosity • The surface free energy term correlates with viscosity. Thus nucleation should require less supersaturation for aqueous solutions than silicate melts. o Among silicate melts, nucleation should occur more readily in basaltic ones, which have low viscosities, than in rhyolitic ones, which have high viscosities. This is what one observes. • Also, we might expect rapid cooling to lead to greater supersaturation than slow cooling. This is because there is an element of chance involved in formation of a crystal nucleus (the chance of bringing enough of the necessary components together in the liquid so that r exceeds rcrit). Nucleation Rate • The first step in is the formation of small clusters of atoms of the right composition. These so-called heterophase fluctuations arise because of statistical fluctuations in the distribution of atoms in the liquid. These fluctuations cause local variations in free energy, and their distribution can be described by the Boltzmann distribution law. The number of clusters of critical size is: N crit = NV e-∆ Gcrit /kT • where Ncrit is the number of clusters of critical size per unit volume containing i atoms, Nv is the number of atoms per unit volume of the cluster, and ∆Gcrit is the total free energy (∆Gtot) of clusters with critical radius (i.e., previous equation with r = rcrit. • For a spherical cluster, this is: ∆ Gcrit 16p s 3V = 3 ∆ Gxt2 Nucleation Rate • If EA is the energy necessary to attach an additional atom to the cluster, then the probability of this is: P = e- EA /kT • According to transition state theory, the frequency of attempts, ν, to overcome this energy is simply the fundamental frequency, ν = kT/h. The attachment frequency is then the number of atoms adjacent to the cluster, N*, times the number of attempts, times the probability of success: N *n P = N * kT - EA /kT e h • Nucleation rate should be this times number of clusters of critical radius: N *n P = N * kT - EA /kT -∆ Gcrit /kT e e h Diopside Nucleation • Combining preexponential terms into a frequency factor, A, and using ∆G ≈ ∆S∆T where ∆T is the offset from the crystallization T (the temperature overstep), for a spherical nucleus we have: • or I = Ae 2 -16 ps 3 V /(3∆ S∆ T )2 kT I µe -1/∆ T 2 The nucleation rate passes through a maximum. This reflects the 1/T dependence of both exponential terms; the formation and growth of heterophase fluctuations falls with temperature. Heterogeneous Nucleation • Heterogeneous nucleation refers to the nucleation of a phase on a pre-existing one. This occurs when the surface free energy between the nucleating phase and the pre-existing surface is lower than between the nucleating phase and the phase from which it is growing. o Examples: dew, pyx-plag intergrowths. Heterogeneous Nucleation • • Consider, for example, a dew drop on a leaf. The balance of surface forces at the threephase contact is: s a s = s b s + s ab cosq • and • If the interfacial energy between the nucleating phase, β, (the dew) and the surface (σβs) (the leaf) is smaller than that between phase α (air) and the surface (σαs), then the angle of intersection, θ, will be small so as to maximize the interfacial surface area between β and s for a given volume of β. In the limit where σβs ≪ σαs then θ will approach 0 and β will form a film coating the surface. As σβs approaches σαs the nucleating phase will form more spherical droplets. If σβs ≥ σαs then θ will be 90° or greater, and heterogeneous nucleation will not occur. • cosq = s a s - s bs s ab Heterogeneous Nucleation • • In metamorphic reactions, nucleation will necessarily always be heterogeneous. Provided the necessary components of the nucleating phase are available and delivered rapidly enough by fluid transport and diffusion, interfacial energy will dictate where new phases will nucleate, nucleation being favored on phases where the interfacial energy is lowest. (Where transport of components limit growth, however, this may not be the case, as phases will nucleate where the components necessary for growth are available.) Diffusion and heat-flow limited growth • • • Crystals can grow only as rapidly as the necessary chemical components are delivered to their surfaces and heat removed (or added). Where diffusion is not rapid enough to supply these components, diffusion will limit growth. Slow diffusion can change the apparent distribution coefficient, because the crystal “sees” the concentrations in the adjacent boundary layer rather than the average concentrations in the liquid. Thus the crystal may become less depleted in elements excluded from the crystal, and less enriched in elements preferentially incorporated in it, than equilibrium thermodynamics would predict. When crystals grow from a liquid there will be a local increase in temperature due to release of latent heat of fusion, ∆Hm, which will retard crystal growth. In most cases, this is at best a minor effect. The effect is probably more important in prograde metamorphic reactions (e.g., dehydration reactions), which are usually endothermic and hence require a continuous supply of energy to maintain crystal growth. Zoning • Diffusion limits the ability of the interior of a crystal to maintain equilibrium with the magma from which it is precipitating. • This leads to zoning in crystals, apparent under the microscope. • Zoning can record magma history. Zoned pyx and plag in lava.