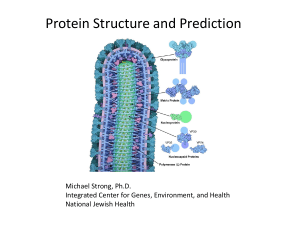
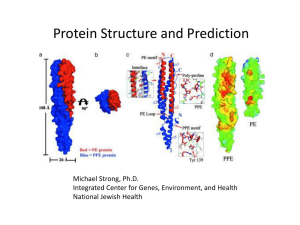
Protein-protein interactions.
advertisement

Protein structure prediction. Protein folds. • Fold definition: two folds are similar if they have a similar arrangement of SSEs (architecture) and connectivity (topology). Sometimes a few SSEs may be missing. • Fold classification: structural similarity between folds is searched using structurestructure comparison algorithms. Protein structure prediction flowchart Protein sequence Does sequence align with a protein of known structure? Database similarity search No Protein family analysis Yes Predicted threedimensional structural model Three-dimensional comparative modeling Yes Threedimensional structural analysis in laboratory No Is there a predicted structure? Yes Relationship to known structure? No Structural analysis From D.W.Mount Protein structure prediction. Prediction of three-dimensional structure of a protein from its sequence. Different approaches: - Homology modeling (query protein has a very close homolog in the structure database). - Fold recognition (query protein can be mapped to template protein with the existing fold). - Ab initio prediction (query protein has a new fold). Homology modeling. Aims to produce protein models with accuracy close to experimental and is used for: - Protein structure prediction - Drug design - Prediction of functionally important sites (active or binding sites) Steps of homology modeling. 1. 2. 3. 4. 5. Template recognition & initial alignment. Backbone generation. Loop modeling. Side-chain modeling. Model optimization. 1. Template recognition. Recognition of similarity between the target and template. Target – protein with unknown structure. Template – protein with known structure. Main difficulty – deciding which template to pick, multiple choices/template structures. Template structure can be found by searching for structures in PDB using pairwise sequence alignment methods. Two zones of protein structure prediction. Sequence identity 100 Homology modeling zone 50 Fold recognition zone 50 100 150 200 Alignment length 2. Backbone generation. If alignment between target and template is ready, copy the backbone coordinates of those template residues that are aligned. If two aligned residues are the same, copy their side chain coordinates as well. 3. Insertions and deletions. insertion AHYATPTTT AH---TPSS deletion Occur mostly between secondary structures, in the loop regions. Loop conformations – difficult to predict. Approaches to loop modeling: - Knowledge-based: search the PDB for loops with known structures - Energy-based: an energy function is used to evaluate the quality of a loop. Energy minimization or Monte Carlo. 4. Side chain modeling. Side chain conformations – rotamers. In similar proteins side chains have similar conformations. If % identity is high - side chain conformations can be copied from template to target. If % identity is not very high modeling of side chains using libraries of rotamers and different rotamers are scored with energy functions. Problem: side chain configurations depend on backbone conformation which is predicted, not real E2 E3 E1 E = min(E1, E2, E3) 5. Model optimization. Energy optimization of entire structure. Since conformation of backbone depends on conformations of side chains and vice versa iteration approach: Predict rotamers Shift in backbone Classwork: Homology modeling. - Go to NCBI Entrez, search for gi461699 Do Blast search against PDB Repeat the same for gi60494508 Compare the results Fold recognition. Unsolved problem: direct prediction of protein structure from the physico-chemical principles. Solved problem: to recognize, which of known folds are similar to the fold of unknown protein. Fold recognition is based on observations/assumptions: - The overall number of different protein folds is limited (1000-3000 folds) - The native protein structure is in its ground state (minimum energy) Fold recognition. Goal: to find protein with known structure which best matches a given sequence. Since similarity between target and the closest template is not high, pairwise sequence alignment methods fail. Solution: threading – sequence-structure alignment method. Threading – method for structure prediction. Sequence-structure alignment, target sequence is compared to all structural templates from the database. Requires: - Alignment method (dynamic programming, Monte Carlo,…) - Scoring function, which yields relative score for each alternative alignment Protein structure prediction: target sequence is compared to structures using sequencestructure alignment Structural templates Score1 Score2 Target sequence Concept of threading: D. Jones et al, 1993 Score3 Protein structure prediction: target sequence is compared to structures using sequencestructure alignment Structural templates Score1 Score2 Target sequence Score3>Score2>Score1 Score3 Structural model of target Scoring function for threading. • Contact-based scoring function depends on amino acid types of two residues and distance between them. • Sequence-sequence alignment scoring function does not depend on the distance between two residues. • If distance between two nonadjacent residues in the template is less than 8 Å, these residues make a contact. Scoring function for threading. Ala Trp Tyr Ile S N w(a , a i , j 1 i j ); S w( Ala, Tyr) w( Ile, Trp) “w” is calculated from the frequency of amino acid contacts in PDB; ai – amino acid type of target sequence aligned with the position “i” of the template; N- number of contacts Classwork: calculate the score for target sequence “ATPIIGGLPY” aligned to template structure which is defined by the contact matrix. A T 1 1 2 3 4 * 5 6 * 7 8 9 10 * Y 2 3 I G * * 6 7 * * * 8 * 9 10 T P Y I G L -0.2 -0.1 0 -0.1 0.5 -0.2 0.2 0.3 -0.1 -0.2 -0.3 0.1 0 -0.2 -0.4 -0.1 0.1 -0.2 -0.4 -0.2 -0.1 -0.2 0.3 0.2 0.4 0.4 0.2 * 4 5 P A * * L 0.3 Optimize the Sum of Residue-Residue Contact Potentials ... …. by a Monte Carlo Alignment Algorithm Evaluation of quality of structural model • Correct bond length and bond angles >> 3.8 Angstroms • Correct placement of functionally important sites • Prediction of global topology, not partial alignment (minimum number of gaps) Success and limitations of structure prediction Limitations: Success: • • Accuracy scores almost doubled from CASP1 to CASP6, might be because of database size Models of small targets are very accurate • • • Models of large and remotely related proteins are not very accurate Domain boundaries are difficult to define Models often do not provide details for functional annotation Adapted from Kryshtafovych et al 2005 GenThreader http://bioinf.cs.ucl.ac.uk/psipred. 1. Predicts secondary structures for target sequence. 2. Makes sequence profiles (PSSMs) for each template sequence. 3. Uses threading scoring function to find the best matching profile. Protein-protein interactions. Common properties of protein-protein interactions. rim • Majority of protein complexes have a buried surface area ~1600±400 Ǻ^2 (“standard size” patch). • Complexes of “standard size” do not involve large conformational changes while large complexes do. core Top molecule • Protein recognition site consists of a completely buried core and a partially accessible rim. • Trp and Tyr are abundant in the core, but Ser and Thr, Lys and Glu are particularly disfavored. Bottom molecule Different types of protein-protein interactions. • Permanent and transient. • External are between different chains; internal are within the same chain. • Homo- and hetero-oligomers depending on the similarity between interacting subunits. • Interface type can be predicted from amino acid composition (Ofran and Rost 2003). Experimental methods Verification of experimental protein-protein interactions. • Protein localization method. • Expression profile reliability method. • Paralogous verification method. Protein localization method. Sprinzak, Sattath, Margalit, J Mol Biol, 2003 A – A3: Y2H B: physical methods C: genetics E: immunological True positives: - Proteins which are localized in the same cellular compartment - Proteins with a common cellular role Expression profile reliability method. Deane, C. M. (2002) Mol. Cell. Proteomics 1: 349-356 Paralogous verification method. PVM method is based on observation that if two proteins interact, their paralogs would interact. Calculates the number of interactions between two families of paralogous proteins. Deane, C. M. (2002) Mol. Cell. Proteomics 1: 349-356 Interaction databases • Experiment (E) • Structure detail (S) • Predicted – Physical (P) – Functional (F) • Curated (C) • Homology modeling (H) • *IMEx consortium Protein interaction databases • Protein-protein interaction databases • Domain-domain interaction databases DIP database • Documents proteinprotein interactions from experiment – Y2H, protein microarrays, TAP/MS, PDB • 55,733 interactions between 19,053 proteins from 110 organisms. Organisms # proteins # interactions Fruit fly 7052 20,988 H. pylori 710 1425 Human 916 1407 E. coli 1831 7408 C. elegans 2638 4030 Yeast 4921 18,225 Others 985 401 DIP database Duan et al., Mol Cell Proteomics, 2002 • Assess quality – Via proteins: PVM, EPR – Via domains: DPV • Search by BLAST or identifiers / text BIND database Alfarano et al., Nucleic Acids Res, 2005 • Records experimental interaction data • 83,517 protein-protein interactions • 204,468 total interactions • Includes small molecules, NAs, complexes Classwork. • Go to DIP webpage (http://dip.doembi.ucla.edu) • Retrieve all interactions for cytochrome C, tubulin, RNA-polymerase from yeast • How many of them are confirmed by several experimental methods? Protein interaction databases • Protein-protein interaction databases • Domain-domain interaction databases InterDom database Ng et al., Nucleic Acids Res, 2003 • Predicts domain interactions (~30000) from PPIs • Data sources: – – – – Domain fusions PPI from DIP Protein complexes Literature • Scores interactions Pibase database • Records domain interactions from PDB and PQS • Domains defined with SCOP and CATH • All inter-domain and inter-chain distances within 6 Ǻ are considered interacting domains • From interacting domain pairs, create list of interfaces with buried solvent accessible area > 300 Ǻ2 Classwork. • Go to Pibase website http://salilab.org/pibase • Select largest structural complexes, 1k73, 1i6h • Compare two complexes in terms of the number of interacting domains, #interactions per node NCBI CBM database Shoemaker et al., Protein Sci, 2006. • CBM – database of interacting structural domains exhibiting Conserved Binding Modes • To retrieve interactions: – Record interactions – Use VAST structural alignments to compare binding surfaces – Study recurring domaindomain interactions Definition of CBM • Interacting domain pair – if at least 5 residue-residue contacts between domains (contacts – distance of less than 8 Ǻ) • Structure-structure alignments between all proteins corresponding to a given pair of interacting domains • Clustering of interface similarity, those with >50% equivalently aligned positions are clustered together • Clusters with more than 2 entries define conserved binding mode. Number of interacting pairs and binding modes • 833 conserved interaction types • 1,798 total domain interaction types • Up to 24 CBMs per interaction type CBM Structures Species 1 154 Jawed vertebrates 2 112 Jawed vertebrates 3 17 Clam,earthworm 4 4 lamprey 5 4 V.stercoraria 6 2 Rice,soybeans 7 2 human 8 2 lamprey • Classify complicated domain pairs by CBMs • Globin example: – 630 pairs – 2 CBMs account for majority Classwork. • Retrieve structures 1GY3, 1E9H, 1OL2 • Examine all interactions within and between chains/domains. • How many CBMs do you find?