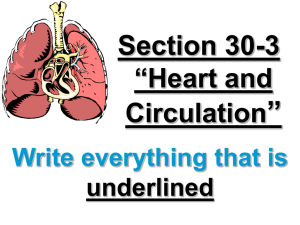
V d
advertisement

Pharmacokinetics Pawitra Pulbutr B.Pharm (Hon.), M.Sc. In Pharm (Pharmacology) Objectives อธิบายถึงปัจจัยทางเคมี-ฟิ สิกส์ท่มี ีผลต่อการส่งผ่านยาผ่านเซลล์เมมเบรน และวิธกี ารที่ยาผ่านเซลล์เมมเบรน อธิบายการจับของยาเข้ากับพลาสมาโปรตีน อธิบายหลักการดูดซึมยา วิธกี ารให้ยาในรูปแบบต่างๆ และปัจจัยที่มีผลต่อการดูดซึมยา อธิบายหลักการกระจายยา ปัจจัยที่มีผลต่อการกระจายยา อธิบายหลักการกาจัดยาออกจากร่างกาย กระบวนการกาจัดยาออกจากร่างกายทัง้ การเปลี่ยนแปลงยา และการ ขับยาออกจากร่างกายรวมทัง้ ปัจจัยที่มีผลต่อการเปลี่ยนแปลงยา อธิบายกระบวนการทางชีวเคมีท่ใี ช้ในการเปลี่ยนแปลงยาในร่างกายทัง้ ใน Phase I และ Phase II reaction อธิบายกลไกการขับยาออกโดยไตในรูปแบบต่างๆ มีความเข้าใจหลักการพื้นฐานของ Clinical pharmacokinetics มีความเข้าใจถึง clearance, volume of distribution และ bioavailability สามารถใช้หลักการทาง Clinical pharmacokinetics ในการคานวณขนาดยาที่เหมาะสม ในเบื้องต้นได้ Pharmacokinetics What body does to the drugs Distribution Absorption กระบวนการที่รา่ งกายจัดการกับยา Metabolism Biotransformation elimination Excretion Locus of action “receptors” Bound Tissue Reservoirs free Free Bound Distribution Systemic circulation Absorption Free drug Bound drug Excretion metabolites Biotransformation Schematic representation of the interrelationship of the absorption, distribution, binding, biotransformation, and excretion of drug and its locus of action How drugs pass the membrane? • Directly diffusion through lipid bilayer • Diffusion through aqueous pore • Carrier protein transport • Pinocytosis ( Cell drinking) Henderson-Hansselbalch equation Weak base pKa = pH + log [BH+] / [B] pH + log[AH] / [A-] Weak acid pKa = Indicates whether weak acid/ base drugs can pass through lipid membrane at different pH medium. Binding of drugs to plasma protein Free drug + Protein Active form Plasma protein 1. Albumin 2. Alpha1 acid glycoprotein 3. Etc:- Beta globulin Bound drugs Inactive form Drug absorption Site of administration Absorption Systemic circulation/ Plasma Site of action Route of administration Oral Parenteral …. Out of GI tract Injection: intravenous (IV), intramuscular (IM), subcutaneous (SC), intraarterial (IA), intrathecal (IT), intraperitoneal (IP) Sublingual (SL) Rectal Inhalation Topical First pass metabolism GI absorption Portal circulation Systemic circulation Drug degradation/ metabolism by GI enzyme Drug degradation/ metabolism by liver enzyme Reduced amount of drug that reach to systemic circulation Oral Degradation GI tract Portal circulation Metabolism Systemic circulation First pass metabolism Pre-systemic metabolism Bioavailability สัดส่วนของยาที่เข้าสู่ systemic circulation First pass metabolism or Presystemic circulation Intestine Liver Reduced Bioavailability Amount of drug administered or amount of drug absorbed may (usually) not be equal to amount of drug in systemic circulation Volume of distribution (Vd) The volume of fluid required to contain the total amount, Q, of drug in the body at the same concentration as that present in the plasma, Cp Vd = Q/ Cp • Vd = Apparent volume • Vd indicates drug distribution • Higher Vd… Higher drug distribution • Vd may be higher than TBW peripheral plasma Low Vd peripheral plasma High Vd Drug Elimination Drug metabolism Drug excretion • Major route of drug elimination • Kidney, Hepatobiliary, Lung, saliva, sweat, milk Why metabolism? What for ?.... To make more water soluble substance Excretion • Major route of drug elimination • Kidney, Hepatobiliary, Lung, saliva, sweat, milk Drug metabolism 1. Phase I reaction Oxidation, Reduction, Hydrolysis Addition of reactive groups or expose masked functional group More chemically reactive 2. Phase II reaction Conjugation reaction Produced inactive and more water soluble compound Drug excretion • Major excretory organ= Kidney (Via urine) • Other route= feces, milk, saliva, exhaled air Renal excretion Glomerular filtration Tubular secretion Tubular reabsorption Biliary excretion & Enterohepatic circulation Drugs Excrete with feces Bile Intestine • Carrier systems • Enterohepatic circulation Reabsorption Drug conjugate • Prolonged drug effect Basic Clinical Pharmacokinetics Pharmacological or Toxic response Dose Drug level in plasma/ blood Effect Pharmacokinetic parameter Clearance (CL) Volume of distribution (Vd) Bioavailability Pharmacokinetics model Single compartment model All parts of body are the same Two compartment model Central (plasma) compartment & Peripheral compartment Multiple compartment model Different parts of body are different Single compartment model In Two compartment model Out Distribute In Out Clearance (CL) อัตราการกาจัดยาใน route นั้นๆ เมื่อเทียบกับความเข้มข้นของยาในเลือด CL organ = Rate of elimination (organ) Concentration of drug in blood (C) CLsystemic = หน่ วยของ CL = CL renal+ CL liver +CL other Volume / time eg. ml/min, L/hr ปริมาตรของ biological fluid ที่ถกู กาจัดยาออกไปใน หน่ วยเวลา 1 First order kinetics • กาจัดยา 50% คงที่ • เป็ นสัดส่วนโดยตรงกับ ความเข้มข้น • ยาส่วนมาก • ที่ความเข้มข้น 25 ยาถูกกาจัด 12.5 • ที่ความเข้มข้น 50 ยาถูกกาจัด 25 • ที่ความเข้มข้น 100 ยาถูกกาจัด 50 Zero order kinetics = Saturation kinetics • การกาจัดยา ปริมาณคงที่ 50 • ไม่ข้ ึนกับความเข้มข้น • การกาจัดยาอิม่ ตัว • ที่ความเข้มข้น 50 กาจัด 50 • ที่ความเข้มข้น 100 กาจัด 50 • ที่ความเข้มข้น 200 กาจัด 50 • ยาบางชนิ ด เช่น phenytoin, ethanol, aspirin At time 0; C0 = Q/ Vd At time t Drug concentration decrease by elimination; follow first order kinetics Ct ln Ct = Co exp - CL/Vd x t = ln Co - CL/Vd x t Plot Ct VS t…. Plasma concentration VS Time curve Figure 3.6 Predicted behavior of single compartment model following IV drug administration ln Ct = ln Co - CL/Vd x t Y = a + bX Slope = -CL/ Vd Half-life = t1/2 ระยะเวลาที่ Ct ลดลงเหลือครึ่งหนึ่ ง =0.693/ Ke Single IV dose … No Absorption needed B C • At steady state concentration (Css) • Rate of infusion = Rate of elimination • Fluctuation = การเปลี่ยนแปลงขึ้นลงของระดับยาใน plasma Interval คือระยะห่างของการให้ยา/ ช่วงของการให้ยา Longer interval … More fluctuation 3-5 half-life = C >B Time to reach CSS Two compartment models Ka Ke = Kmet + Kexc Distribution Elimination Kinetic of diazepam elimination in man following a single dose in two compartment model Distribution Bioavailability สัดส่วนของยาที่สามารถเข้าสู่ systemic circulation Fraction (F) of dose given that could reach systemic circulation Consider both Rate and Extent Available dose = F x dose ที่ได้รบั จริง ที่ให้ Cmax Rate Extent Tmax A & B C Extent A>C>B AUC = Area under the plasma concentration time curve = Amount of drug that reach to sys. circulation MTC Therapeutic concentration MEC duration onset Consider A • MEC = Minimum effective concentration • MTC = Minimum toxic concentration/ Maximum tolerated concentration Design and Optimization of Dosage Regimens Target level = ระดับความเข้มข้นของยาที่ตอ้ งการ Drugs that should design target level Concentration - response relationship Narrow therapeutic index High toxicity Effect can not be evaluated easily To be able to determine drug level Digoxin, Theophylline, Aminoglycosides, etc. Maintenance dose (MD) At CSS ขนาดยาที่ให้เพือ่ คงระดับยาให้อยู่ท่ี CSS Rate in = Rate out Dosing rate = Rate of elimination CL = Rate of elimination Cp Dosing rate = CL x CSS = CL x TC Target concentration Consider bioavailability, F Available dose (ที่ได้รบั จริง) = Dose (ที่ให้) x F Dose (ที่ให้) = Available dose / F MD =Dosing rate = CL x CSS = CL x TC F F For intermittent dose การให้ยาเป็ นช่วง เช่นทุก......ชัว่ โมง MD = Dosing rate x Dosing interval (mg/hr) (hr) Loading dose (LD) ขนาดยาที่ให้เพือ่ ให้ได้ระดับยาที่ target concentration ที่ตอ้ งการอย่างรวดเร็ว จาก Vd = Q / Cp หาก Cp คือTarget concentration ที่ตอ้ งการ (desired concentration) … (ซึ่งอาจเป็ น Css) Q คือ Amount of drug ที่จะให้ target concentration ที่ตอ้ งการ Q = LD LD = Vd x TC การให้ loading dose ต้องคานึ งถึง Rate of administration ด้วยว่าจะก่อให้เกิด toxicity หรือไม่ Pharmacokinetics What did you get?